





Abstract
Chronic obstructive pulmonary disease (COPD) is a known risk factor for developing lung cancer but the underlying mechanisms remain unknown. We hypothesise that the COPD stroma contains molecular mechanisms supporting tumourigenesis.
We conducted an unbiased multi-omic analysis to identify gene expression patterns that distinguish COPD stroma in patients with or without lung cancer. We obtained lung tissue from patients with COPD and lung cancer (tumour and adjacent non-malignant tissue) and those with COPD without lung cancer for profiling of proteomic and mRNA (both cytoplasmic and polyribosomal). We used the Joint and Individual Variation Explained (JIVE) method to integrate and analyse across the three datasets.
JIVE identified eight latent patterns that robustly distinguished and separated the three groups of tissue samples (tumour, adjacent and control). Predictive variables that associated with the tumour, compared to adjacent stroma, were mainly represented in the transcriptomic data, whereas predictive variables associated with adjacent tissue, compared to controls, were represented at the translatomic level. Pathway analysis revealed extracellular matrix and phosphatidylinositol-4,5-bisphosphate 3-kinase–protein kinase B signalling pathways as important signals in the tumour adjacent stroma.
The multi-omic approach distinguishes tumour adjacent stroma in lung cancer and reveals two stromal expression patterns associated with cancer.
Abstract
A multi-omic approach identified gene expression programmes distinguishing COPD lung stroma associated with lung cancer http://ow.ly/j1Da30jWH1X
Introduction
Independent of smoking, chronic obstructive pulmonary disease (COPD) is a known risk factor for the development of lung cancer [1, 2]. This paradigm is further supported by the observation that tumours are more likely to arise in association with areas of emphysema [3, 4]. Central to the pathophysiology of airflow obstruction in COPD is remodelling of lung extracellular matrix (ECM) with loss of elasticity and increased resistance to airflow. This ECM remodelling is accompanied by small airway fibrosis and inflammation, a stromal milieu with the potential to promote tumourigenesis of epithelial cells that have previously incurred oncogenic mutations from exposure to tobacco or other oncogenic stressors.
It is well recognised that cancer initiation occurs as a progression of genetic mutations. However, it is becoming more apparent that tumours are integrally connected with their microenvironment [5, 6]. This microenvironment consists of ECM along with a stroma consisting of fibroblasts, endothelial cells and resident immune cells that can promote tumour progression. These stromal cells do not acquire genetic mutations, nevertheless, it is well accepted that cancer-associated fibroblasts, the primary stromal cell that produces ECM, are a major contributor in tumour progression. Recent evidence also places the fibroblast and ECM at the centre of tumour initiation. Senescent human fibroblasts have been shown to stimulate both malignant and premalignant epithelial cells to proliferate while “normal” epithelial cells remain unaffected [7]. In a murine model of breast cancer, fibroblasts devoid of the tumour suppressor gene PTEN dramatically accelerate cancer initiation, in a manner that is partially dependent on fibroblast secretory factors [8]. Our study sought to identify the specific alterations in the COPD-stroma that promote lung carcinogenesis, as these remain unknown.
The mechanisms creating a pro-malignant stromal environment are likely complex. Although proteins are the effector molecules, their regulation can occur at multiple levels including transcription, translation and post-translational modification. Early on, transcriptional regulation was the major focus of study; however, translation regulation has emerged as a significant mechanism in regards to carcinogenesis [9–11]. Therefore, genome-wide approaches are necessary to understand the complexity of carcinogenesis. In this regard, we sought to understand the role of the COPD lung stroma in lung cancer using a multi-omic approach.
Materials and methods
Study population and lung tissue samples
The University of Minnesota Institutional Review Board Human Subjects Committee approved this study. Surgical lung tissue samples were obtained from the Mayo Clinic Lung Specimen Registry from individuals with COPD who underwent surgical resection for Stage I or II adenocarcinoma (table 1). Tissue samples consisted of tumour and adjacent lung from 32 patients with adenocarcinoma and COPD. Controls were obtained from the National Institutes of Health Lung Tissue Research Consortium (NIH LTRC) and consisted of lung tissue from 32 individuals with COPD without lung cancer (table 1). All samples were obtained using identical procedures as outlined by the NIH LTRC. All subjects, cases and controls were current or former smokers. These lung tissue biopsies are referred to as “tumour, adjacent and control”. In total there were 32 tissue samples with shared transcriptome, translatome and proteomic data (10 controls and 11 cases with one tumour and one adjacent tissue each; table 2). The number of samples that are available for each data type are shown in table 3.
Patient demographics for proteomic data
Patient demographics for shared transcriptomic, translatomic and proteomic data
Sample availability by data type and the overlap of all three data types
Proteomics and RNA sequencing
Lung tissue was processed for isobaric tag for relative and absolute quantitation (iTRAQ) labelling following manufacturers’ recommendations and as previously published [12]. Lung tissue was processed for total and polysomal mRNA isolation and sequencing as previously described [13]. Detailed experimental design information is available in the supplementary material. Protein mass spectrometry data is available at the PRoteomics IDEntifications (PRIDE) database (www.ebi.ac.uk/pride/archive/; accession number PXD007919). RNA sequencing data is available at the Gene Expression Omnibus (GEO) data repository (www.ncbi.nlm.nih.gov/geo/; accession number GSE106899).
Statistical analysis
All data analysis was performed using the R Environment for Statistical Computing (www.r-project.org/; R version 3.3.2). Prior to data integration, data underwent pre-processing and independent screening as described in the supplementary material [14–16]. After pre-processing there were 1486 proteomic, 5989 transcriptome and 8014 translatome genes or proteins available. Next, we determined an integrative composite biomarker for sample status (tumour, adjacent and control) from the three different omic data types (proteomic, transcriptome and translatome) using a multi-step approach described previously [17].
For each data type, we first conducted a univariate linear modelling step for each gene or protein to filter out variables with little predictive power. After filtering, we performed an integrative dimension reduction of the remaining variables using the Joint and Individual Variation Explained (JIVE) method [18, 19]. JIVE is an extension of principal components analysis for multi-omic data and it identifies latent structures shared among and expressed within multiple high-throughput data types. “Joint components” define sample patterns that explain substantial variability across multiple data types and “individual components” are sample patterns that explain substantial variability in one data type but not others [17].
After performing JIVE we used the lung-cancer sample types (tumour, adjacent and control) as our outcomes in a multinomial logistic regression using the JIVE components as predictors. The components were either included or excluded from the model by a stepwise (forward/backward) selection algorithm to minimise the Akaike Information Criteria (AIC) [20]. This gave a composite model to predict the odds of a tissue sample being cancerous, tumour-adjacent, or non-cancerous COPD from all three omics datasets.
To assess the accuracy of the fitted model we performed N-fold (leave one out) cross-validation where a single tissue sample was removed as a test case, the model parameters were estimated using the remaining (n=31) samples as a training set and the prediction of the resulting model was assessed using the test case. This procedure was repeated for each sample as the test case.
To determine which genes or proteins were most influential, the JIVE components were mapped back to the original variable space to give meta-loadings for each variable [17]. These could be interpreted as giving the linear weight (i.e. coefficient) of each variable in the fitted model for each comparison. We performed a pathway analysis using the meta-loadings to determine which biological functions were most over-represented in the fitted model. Those genes which had an absolute meta-loading greater than or equal to the mean of the absolute meta-loading for a given comparison were used as the active gene set for each data type. The WEB-based GEne SeT AnaLysis Toolkit (WebGestalt; www.webgestalt.org/) [21] was used to perform an over-representation enrichment analysis using KEGG pathways as described in the supplementary material.
Results
Samples and data acquisition
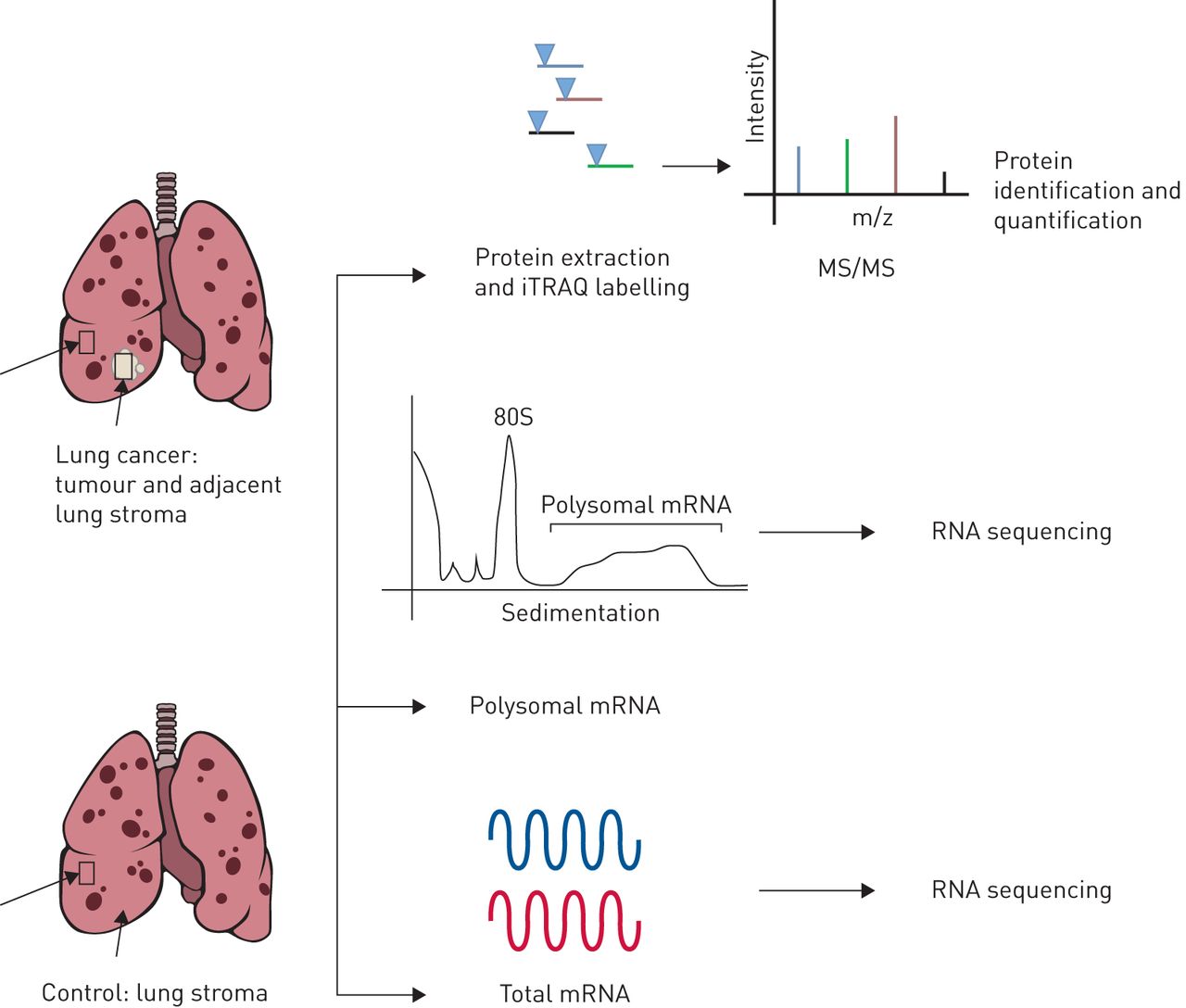
We compared lung tumour and non-cancerous adjacent lung tissue from patients with COPD to tissue from patients with COPD but without lung cancer (figure 1). Proteomic analysis was available for 32 patients with COPD and lung cancer and 32 controls. Lung function was more severe in the non-cancerous control group with an average % predicted forced expiratory volume in 1 s (FEV1) of 18% compared to 53% in those with lung cancer (table 1). Proteomic analysis consisted of iTRAQ-based quantitative mass spectrometry across 16 runs, obtaining an average of 27 432 spectra matched to 12 457 distinct peptides and 2105 inferred proteins at a 1% global false discovery rate (FDR). Twenty-one subjects (11 cases and 10 controls) had samples adequate for both proteomic analysis and mRNA sequencing, both total and polyribosomal (table 2), with an average of 24 484 971 reads and a median read quality of 38. This led to a read mapping percentage of 52.6, identifying an average of 5602 genes per sample from mRNA sequencing of total mRNA (transcriptome) and polysome-associated mRNA (transcripts bound by more than three ribosomes). The latter identifies efficiently translated mRNA, hereafter referred to as the translatome. The lower mapping was attributed to the relatively small sample mass requiring additional amplification.

The multi-omic profiling approach for lung tumour, adjacent non-cancerous lung tissue and lung tissue from chronic obstructive pulmonary disease controls. iTRAQ: isobaric tag for relative and absolute quantitation; MS/MS: tandem mass spectrometry.
Independent screening
Under univariate screening, the translatome data did not yield significant independent biomarker associations with cancer status under the FDR threshold of 0.1. The transcriptome data attained five significant independent biomarker associations with cancer status and the proteomic data attained 1116 significant independent biomarker associations. The larger number of associations detected for the proteomic data is due in part to its much larger sample size (n=96 for proteome versus n=33 for mRNA), which yields more statistical power to detect independent associations under the rigorous multiple testing criteria used here.
Venn diagrams demonstrate the number of significant proteins (FDR <0.1) for each comparison (figure 2). Within the proteomic independent association results, the majority of biomarker associations were significant for comparing tumour samples to paired adjacent samples and also for comparing tumour samples to controls (figure 2b). A smaller number of proteins distinguished the adjacent samples from controls. For the transcriptomic data, one gene (DDIT4) was a shared significant independent biomarker between the cancer status associations of tumour and control as well as adjacent and control (figure 2a). Using a more stringent threshold, FDR <0.05, no transcriptomic associations remained and 314 total proteomic associations remain significant.

Venn diagrams for a) shared transcripts and b) proteins between sample comparisons using pairwise t-tests (false discovery rate (FDR) <0.1).
For the transcriptome data, 35 genes had a significant correlation with % predicted FEV1 within controls, while for the translatome data 18 genes had a significant correlation with % predicted FEV1 within controls (FDR <0.1). These genes had no overlap with those genes that correlate with cancer status in figure 2. No other significant associations were detected with % predicted FEV1 or age for the transcriptome, translatome or proteomic data, either within controls or within cancer patients.
Data integration and predictive modelling identifies eight latent data structures
To integrate these three data sets into a useful predictive model we used the JIVE algorithm for multi-source data [18, 19]. JIVE is an extension of principal components analysis for multi-omic data and identifies latent structures (i.e. components) shared among and expressed within multiple high-throughput data types. Joint components define sample patterns that explain substantial variability across multiple data types and individual components are sample patterns that explain substantial variability in one data type but not others [17].
After filtering by univariate multinomial logistic regression we had 612, 1108 and 574 genes for the proteomic, transcriptome and translatome data, respectively. The number of proteins or genes at each stage of filtering and the overlap between transcriptome and translatome at each stage is given in figure 3. Permutation testing identified two joint JIVE components, zero significant additional individual components for proteomic data, three individual components for the transcriptome and three individual components for the translatome for a total of eight underlying latent patterns. The three sample groups (tumour, adjacent and control) are distinguished by the two joint components, as seen in the top-left panel of figure 4.
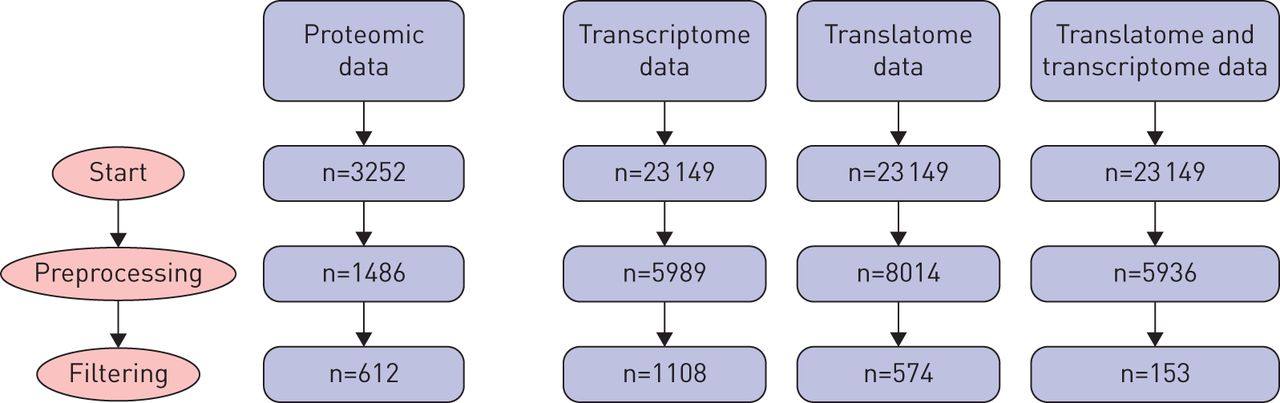
Diagram of the number of genes or proteins at different stages of preprocessing and filtering prior to Joint and Individual Variation Explained (JIVE) analysis. Preprocessing details are available in the supplementary material. The right-hand column gives the number of genes shared by the transcriptome and translatome data after each step.

Scatterplots of joint and individual variation explained (JIVE) scores for two joint components and the first individual components for the transcriptome data and translatome data. Each point represents one lung tissue sample and colors give cancer status. The first joint component shared by the three data types (a-c) shows a separation between non-cancer (control and adjacent) and cancer samples. The second joint component (d, e) shows a distinction between the adjacent samples and the rest of the samples. The individual structures (transcriptome and translatome, respectively) did not show strong differentiation of cancer status (f).
Next, we fit a multinomial logistic regression model to predict sample group using the eight identified JIVE components as predictors. That is, the log odds ratio that a sample belongs to a given group is modelled by a weighted linear combination of the two joint components, three individual components for the transcriptome and three individual components for the translatome. The predicted group probabilities for each sample under N-fold cross-validation are shown in figure 5. The model robustly distinguishes all three sample groups. Using a class prediction probability of 0.5 as a threshold, just three samples were misclassified (90.6% accuracy): a tumour sample was classified as tumour adjacent (10 out of 11 correctly classified), an adjacent sample was misclassified as a control (10 out of 11 correctly classified) and a control sample was misclassified as a tumour (9 out of 10 correctly classified). The log predicted probabilities did not have a significant correlation with % predicted FEV1 or age within control patients or within cancer patients (p>0.05).

Predicted group probabilities for multinomial logistic regression using N-fold cross-validation (n=32).
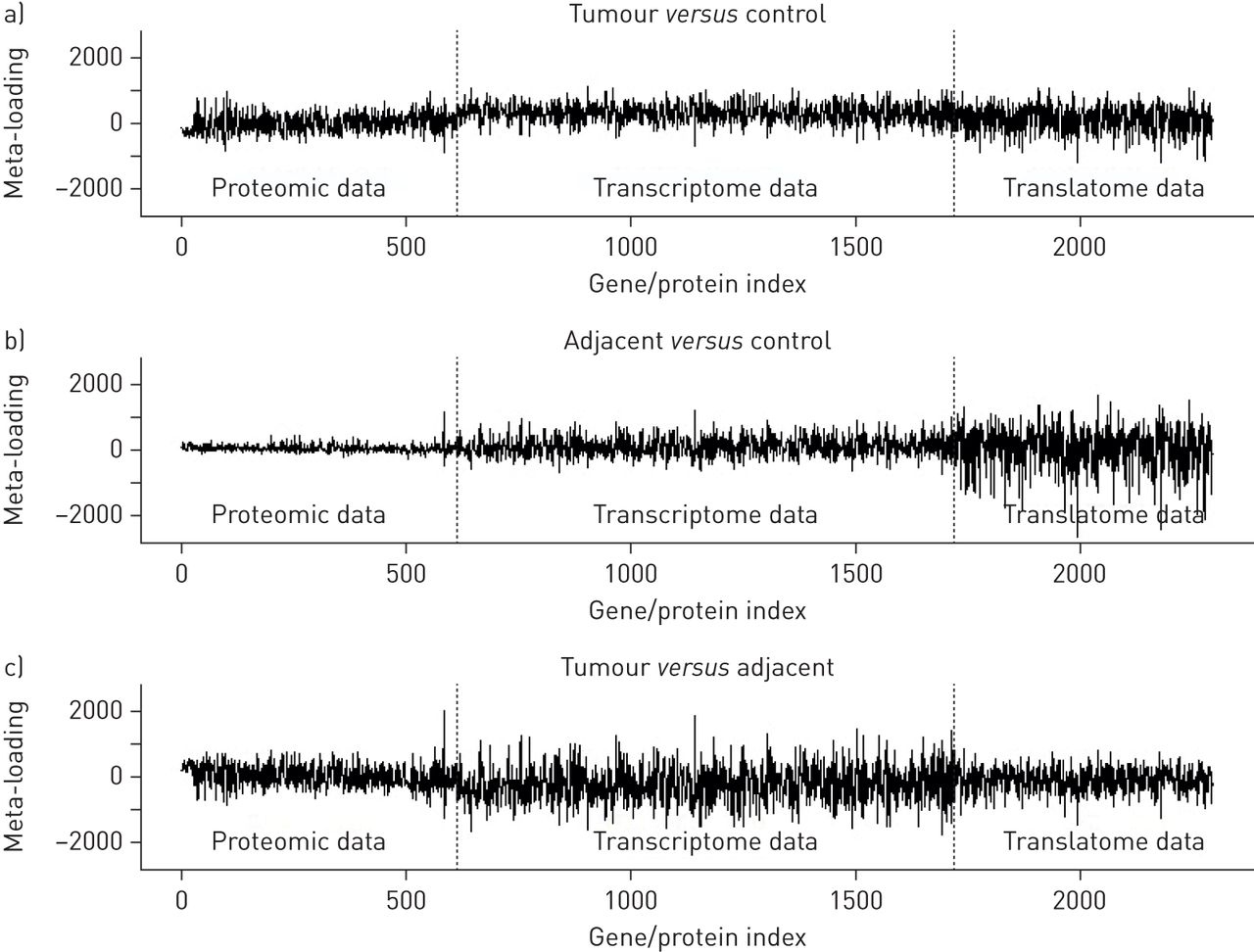
Multinomial logistic regression model (supplementary figure S1) coefficients for the JIVE scores are summarised in supplementary table S1. The resulting gene and protein meta-loadings are shown for each comparison (tumour versus control, adjacent versus control and tumour versus adjacent) in figure 6. These plots show meta-loading against variable (gene or protein) index, revealing the size and direction of each variable's contribution to the predictive model for each comparison. It is apparent that predictive variables for tumours compared to control tissue were evenly distributed across proteomic, transcriptomic and translatomic data. Predictive variables of COPD stroma associated with cancer versus adjacent COPD stroma not associated with cancer were primarily found in translatomic data (polyribosomal-associated mRNA), while predictive variables of tumour versus adjacent were primarily found in transcriptomic data (total mRNA). This pattern suggests that lung tissue adjacent to lung cancers, but not containing lung cancer, display unique signatures that manifest at different levels of regulation. These datasets indicate that a thorough interrogation of not only the proteome, but also the transcriptome and translatome, provide molecular insight that would otherwise be discarded.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Meta-loadings for variables in each of the three data types (proteome, transcriptome and translatome) for each of the sample comparisons considered: a) tumour versus control, b) adjacent versus control and c) tumour versus adjacent.
KEGG pathway analysis
Pathway enrichment analysis of the meta-loadings obtained from the JIVE predictive analysis revealed several significant pathways for each comparison across the three data types (table 4). For the proteome data, six pathways were significant for the tumour versus adjacent comparison and five were significant for the tumour versus control comparison. However, for the translatome data, six pathways were significant for the adjacent versus control comparison. For all other analyses all pathways were insignificant after correction for multiple comparisons (FDR >0.1). These findings are consistent with the translatome being most relevant to the distinction of adjacent versus control samples, while the proteomic data is more relevant for the other two comparisons. However, several pathways are shared among the different comparisons and data types. ECM-receptor interaction, focal adhesion and phosphatidylinositol-4,5-bisphosphate 3-kinase–protein kinase B (PI3K–Akt) signalling were highly significant for each of the three analyses that yielded significant results.
Significantly over-represented pathways (false detection rate (FDR) <0.1) using the average absolute meta-loading threshold
An analogous pathway analysis based on univariate t-statistics rather than JIVE meta-loadings for each dataset and comparison was also performed. The pathways achieving significance were again found only for the three analyses above (tumour versus adjacent for proteomic data, tumour versus control for proteomic data and adjacent versus control for translatome data). The results closely resembled those in table 3 (see supplementary table S2).
Discussion
The relationship between COPD and lung cancer was described over three decades ago [1, 2]; however, the molecular underpinnings of this observation remain elusive. In this study, we utilised a multi-omic approach on lung tissue that included tumour, adjacent non-malignant lung tissue and COPD lung tissue in subjects without lung cancer to uncover pre-malignant signals in lung stroma. This multi-omic approach included quantitative mass spectrometry proteomics along with RNA sequencing (RNA-Seq) on total cytoplasmic mRNA (transcriptomics) and polysomal-associated mRNA (translatomics). To integrate our multi-omic datasets, we utilised the joint and individual variation explained (JIVE) method [18, 19] that simultaneously models components that are shared across all datasets. From these we constructed a multi-omic predictive model that gave statistically robust separation of the three tissue types (i.e. tumour, non-tumour adjacent tissue and control COPD tissue).
Among our datasets the proteomic dataset had the largest sample size due to limitations in the original sample mass. We observed the largest differences in expression when comparing tumour and non-tumour tissues, either adjacent tissue or that from COPD controls. This is not surprising considering cancers replace normal lung structure creating a unique molecular background. Interestingly, we found significant changes between stroma proteome from tissue adjacent to the lung cancer compared to control tissue, i.e. that not associated with lung cancer.
To explore and integrate the three multi-omic datasets we utilised the JIVE algorithm after filtering by univariate multinomial logistic regression. The JIVE algorithm reduced the large dataset variables to eight latent patterns. The three sample groups (tumour, adjacent and control) were illustrated most by two joint components. Six additional individual components or patterns were identified for the transcriptome and translatome (three each). Using these components, we generated a model to predict sample group identity, which robustly distinguished and separated all three sample groups. Next, we determined the size and direction of each variable's contribution to the model. Predictive variables of tumour compared to adjacent lung stroma were found primarily in the transcriptomic data set. This is not surprising since nonsmall cell lung cancer (NSCLC) consists of epithelial cells that have undergone genetic alterations often manifesting in alterations in transcription [22]. However, the predictive variables of non-malignant stroma, i.e. tumour adjacent stroma versus control stroma, were manifested in the polyribosomal mRNA dataset or translatome [23], suggesting regulation occurs at the level of translation [24, 25]. These findings are consistent with other reports that gene expression in cancer initiation and progression is controlled at the level of translational [26–28].
Pathway analysis of the genes demonstrating changes in the adjacent tissue translatome identified ECM and PI3K–Akt signalling as potential pre-malignant pathways. Both ECM and PI3K–Akt over-representation were common in the tumour and adjacent tissue, but differed from controls. Changes in the proteome were also related to the ECM and the PI3K–Akt signalling pathways, but these differences indicated over-representation in the tumour tissue relative to adjacent tissue and controls. This suggests that abnormal ECM and PI3K–Akt play a role as pre-malignant signals.
Translation is a tightly regulated process and dysregulation of this process is associated with the development and progression of many cancers, including lung cancer [26]. The eukaryotic translation initiation factor 4E (eIF4E) forms a complex that enables translation. Under homeostatic conditions, eIF4E is sequestered, however, stimulation via the PI3K–Akt–mammalian target of rapamycin (mTOR) pathway leads to assembly of the eIF4E complex and increased translation [29]. Elevated levels of eIF4E are associated with both experimental and human cancers, including lung cancer [26, 29]. Our data was consistent with these findings, as the tumour samples demonstrated increased representation of the PI3K–Akt signalling pathway compared to both adjacent tissue and controls. However, the role of eIF4E in pre-malignant lesions is less defined. Li et al. [28] identified eIF4E positive cells in adjacent, non-malignant tissues in association with oral squamous cell cancer. We identified the PI3K–Akt signalling pathway to be significantly over-represented in tissue adjacent to the tumour compared to control stroma and this pattern was expressed primarily in the translatome or polyribosomal fraction. Thus, lung stroma that is histologically free of lung cancer has pre-malignant signals of increased translation via the PI3K–Akt signalling pathway.
The role of AKT in COPD is less well known. In a study by Hosgood et al. [30], a single nucleotide polymorphism (SNP) of the PTEN gene was identified that associated with COPD. PTEN is an important regulator of cell-cycle progression and cellular survival and also functions as a negative regulator of AKT. Loss of PTEN is associated with cancer initiation and therefore it is designated as a tumour suppressor gene. In a murine model of breast cancer, loss of PTEN in stromal fibroblasts dramatically accelerated both cancer initiation and progression [8]. These findings suggest that AKT regulation may be the link between COPD and cancer initiation.
The tumour stroma consists of non-malignant cells, such as fibroblasts, mesenchymal cells and immune cells along with vasculature and ECM. In the last two decades, it has become apparent that the tumour stroma plays an active role in tumour cell recruitment, growth and spread [6, 31, 32]. However, the role of the ECM as a pre-malignant signal remains less clear. Abnormal ECM remains a hallmark of COPD since the discovery of elevated elastolytic and other related proteases that disrupt the ECM in COPD [33, 34]. These matrix-degrading proteases are produced by non-malignant cells, but once released they can be wielded by malignant or pre-malignant cells to aid micro-invasiveness or activation of signalling pathways [35]. As expected we found evidence of ECM differentiation between tumour and adjacent tissue, but also found differences when comparing tumour adjacent non-malignant stroma and COPD lung tissue not associated with cancer. These findings are tempered by the difference in COPD severity between controls and those with cancer, but highlights the important and somewhat covert role ECM likely has in both COPD and the pre-malignant stroma.
A limitation of our study is the difference in COPD severity in lung cancer associated COPD versus controls (i.e. controls had more severe COPD relative to those with lung cancer). Since our dataset was not large enough to control for COPD severity, we cannot determine with certainty whether some of the differences observed are related to COPD severity. However, the same pathways (e.g. ECM and PI3K–Akt) were represented in the distinction of tumour tissue from matched adjacent tissue, which suggests they are related to carcinogenesis and not COPD severity. Moreover, the identified genes and the predictions from the multinomial model did not correlate with COPD severity within the control or cancer cohorts. Although our adjacent non-malignant lung tissue did not contain histological evidence of malignancy, we cannot guarantee that the tumour influenced stromal phenotype at a distance through secreted factors. In addition, we cannot guarantee that the control group did not carry pre-malignant changes that would manifest at a later time point. However, if true this would introduce noise and weaken our statistical analysis, which is not suggested by the robust distinctions found in our study.
In conclusion, we set out to explore the COPD cancer-associated stroma for molecular signals supporting tumourigenesis. We found that COPD lung tissue adjacent to, but not containing, adenocarcinoma expresses distinct molecular signals compared to both tumour and non-malignant COPD tissue (the control). Specifically, molecular signals in tumour-associated tissue were manifested at the level of translation and were enriched for ECM and PI3K–Akt signalling. This suggests abnormal ECM and PI3K–Akt signalling as potential pre-malignant pathways.
Supplementary material
Supplementary Material
Please note: supplementary material is not edited by the Editorial Office, and is uploaded as it has been supplied by the author.
Supplementary methods and tables ERJ-02665-2017_Supplement
Supplementary figure S1. The multinomial logistic regression model was fit to predict cancer status from the eight JIVE components. ERJ-02665-2017_Figure_S1
{kind=link}
Footnotes
This article has supplementary material available from erj.ersjournals.com
Conflict of interest: None declared.
Support statement: This research was supported by the National Heart, Lung and Blood Institute (NHLBI) grant R01-HL107612. Funding information for this article has been deposited with the Crossref Funder Registry. This material is the result of work supported with resources and the use of facilities at the Minneapolis VAMC. The contents do not represent the views of the U.S. Department of Veterans Affairs or the United States Government.
- Received December 20, 2017.
- Accepted May 6, 2018.
- The content of this work is not subject to copyright. Design and branding are copyright ©ERS 2018
References










