





Abstract
Pulmonary arterial hypertension (PAH) is characterised by excessive pulmonary vascular remodelling involving deregulated proliferation of cells in intima, media as well as adventitia. Pulmonary arterial endothelial cell (PAEC) hyperproliferation and survival underlies the endothelial pathobiology of the disease.
The indispensable involvement of Notch1 in the arterial endothelial phenotype and angiogenesis provides intriguing prospects for its involvement in the pathogenesis of PAH.
We observed an increased expression of Notch1 in lungs of idiopathic PAH (IPAH) patients and hypoxia/SU5416 (SUHx) rats compared with healthy subjects. In vitro loss- and gain-of-function studies demonstrated that Notch1 increased proliferation of human PAECs (hPAECs) via downregulation of p21 and inhibited apoptosis via Bcl-2 and Survivin. Inhibition of Notch signalling using the γ-secretase inhibitor dibenzazepine dose-dependently decreased proliferation and migration of hPAECs. Notably, Notch1 expression and transcriptional activity were increased under hypoxia in hPAECs and knockdown of Notch1 inhibited hypoxia-induced proliferation of the cells. Furthermore, in vivo treatment with a γ-secretase inhibitor (AMG2008827) significantly reduced the right ventricular systolic pressure and right heart hypertrophy in SUHx rats.
Here, we conclude that Notch1 plays a critical role in PAH and Notch inhibitors may be a promising therapeutic option for PAH.
Abstract
Increased Notch1 regulates endothelial proliferation, migration and survival in PAH, making it a novel target http://ow.ly/Z7iD3008Dxn
Introduction
Pulmonary arterial hypertension (PAH) is a progressive disease characterised by increased pulmonary vascular resistance (PVR) leading to right heart hypertrophy and ultimately death of the patient due to right heart failure. Pathogenesis of PAH involves a complex and multifactorial process in which endothelial cell dysfunction appears to play an integral role in mediating the structural changes in the pulmonary vasculature [1]. Direct evidence in support of a proliferative endothelial cell population resistant to apoptosis in vivo has been provided by the detection of increased expression of various proliferation markers (Ki-67/MIB-1) [2] and angiogenesis and survival-related molecules [3, 4], such as vascular endothelial growth factor (VEGF) and VEGF receptor (VEGFR)-2 as well as hypoxia-inducible factor (HIF)-1α and -1β [4]. Endothelial cells have further been shown to harbour mutations in the Bax gene, along with decreased expression of Bax and caspase-3 [5, 6]. Additionally, endothelial cells isolated from idiopathic PAH (IPAH) patients show increased proliferation in response to various growth factors in vitro [5]. Thus, targeting the highly proliferative and apoptosis-resistant endothelial cells could provide a therapeutic intervention for PAH.
Notch signalling has been implicated in vascular development, homeostasis and injury response [7–9]. Notch receptors (Notch1–4) are transmembrane proteins, displayed on the cell surface as heterodimers. Interaction between Notch receptors and ligands (Jagged or Delta-like ligands (DLL)) expressed on adjacent cell membranes results in activation of signalling. Binding of the ligand triggers two proteolytic cleavages in Notch receptor. Second cleavage mediated by γ-secretase releases Notch intracellular domain (NICD), which then translocates to the nucleus. In the nucleus, NICD binds to the DNA-binding protein CSL, also called recombination signal sequence binding protein Jκ (RBP-Jκ), leading to transcription of Notch target genes, which in turn control numerous cellular processes, such as stem cell maintenance, cell fate specification, differentiation, proliferation and apoptosis [10].
Previously, Li et al. [11] reported increased activation of Notch3 signalling in pulmonary artery smooth muscle cells (PASMCs) from PAH patients, and mutations of Notch3 have been identified in patients with PAH and shown to promote cell proliferation and viability [12]. However, several studies have also linked Notch1 signalling to vascular development and injury. Notch1 knockout mice resulted in embryonic lethality due to defects in somitogenesis and cardiovascular abnormalities [13]. Cre-mediated deletion of endothelial Notch1 during embryonic development was also lethal, proving that arrested growth is associated with loss of the gene in vessels/endocardium. Decreased neointima formation in heterozygous Notch1+/− mice [14] and Hey2−/− [15] mice after carotid artery ligation provided strong evidence for its role in response to vascular injury. Several reports have further linked Notch1 to endothelial proliferation [16–19]. Although well characterised in the systemic vasculature [7] and vascular injury [8, 9], Notch1 is poorly understood in the pulmonary vasculature. This led us to investigate the expression and potential contribution of Notch1 signalling in the pathogenesis of PAH. Specifically, we aimed to: 1) profile Notch1 expression in lungs from patients with IPAH or from hypoxia/SU5416 (SUHx) rats, 2) determine Notch1 expression and function under hypoxia in human PAECs (hPAECs), 3) investigate the effect of Notch1 gain- and loss-of-function on proliferation and apoptosis of hPAECs, and 4) investigate the effect of a pan Notch inhibitor (γ-secretase inhibitor) on hPAECs in vitro and SUHx rats in vivo.
Materials and methods
Primary cells and cell culture
hPAECs were purchased from Lonza (Basel, Switzerland).
Plasmids and small interfering RNA
pFLAG-CMV2-Notch1 intracellular domain (Notch1ICD) plasmid was a kind gift from Professor Cheol O. Joe (Korea Advanced Institute of Science and Technology, Daejeon, South Korea). Notch1 promoter plasmid was a kind gift from Professor G. Paolo Dotto (University of Lausanne, Lausanne, Switzerland). Small interfering RNA (siRNA) targeting Notch1 mRNA (si-Notch1) and scrambled siRNA (si-Scr) were purchased from SABiosciences (Frederick, MD, USA).
RNA isolation and real-time reverse transcriptase PCR
Total RNA from tissues and cells was transcribed using an ImProm-II Reverse Transcription System (Promega, Madison, WI, USA), followed by real-time PCR analysis of various genes using the primers described in online supplementary table S2.
Western blotting analysis
Tissues and hPAECs were lysed in radioimmunoprecipitation assay lysis buffer (Thermo Scientific, Waltham, MA, USA) and the lysates were subjected to Western blots using various antibodies (online supplementary table S3).
Assessment of proliferation and apoptosis of hPAECs
Proliferation and apoptosis of hPAECs was assessed using a bromodeoxyuridine incorporation assay (Roche, Basel, Switzerland) and in situ cell death detection kit (Roche) according to the manufacturer's instructions. The Bax/Bcl-2 ratio was used to assess the degree of apoptosis.
Assessment of migration and tube formation of hPAECs
Migration of hPAECs was assessed using a modified transwell Boyden chamber, while tube formation assay was carried out using the Matrigel™ assay.
Luciferase assay
Human embryonic kidney (HEK) cells were transfected with the Notch1 promoter/reporter gene construct followed by hypoxia exposure (1% O2) and measured for luciferase activity using a Dual Luciferase Reporter Assay (Promega, Madison, WI, USA).
Immunohistochemistry
Paraffin-embedded tissue sections were stained for Notch1 using a NovaRED™ horseradish peroxidase substrate kit (Vector, Burlingame, CA, USA) according to the manufacturer's instructions. Serial section staining with von Willebrand factor (an endothelial marker) and α-smooth muscle actin (α-actin; a smooth muscle marker) was additionally carried out.
Echocardiography, haemodynamic and right ventricular hypertrophy measurements
Transthoracic echocardiography, haemodynamics and tissue preparations were performed as described previously [20], and are described in detail in the online supplementary material.
Statistical analysis
Data are presented as mean±sem. Unless otherwise stated, statistical comparisons of samples were performed by one-way ANOVA followed by Dunnett's post hoc test. Two samples were compared using the t-test.
Results
Notch1 expression and localisation in IPAH and donor lungs
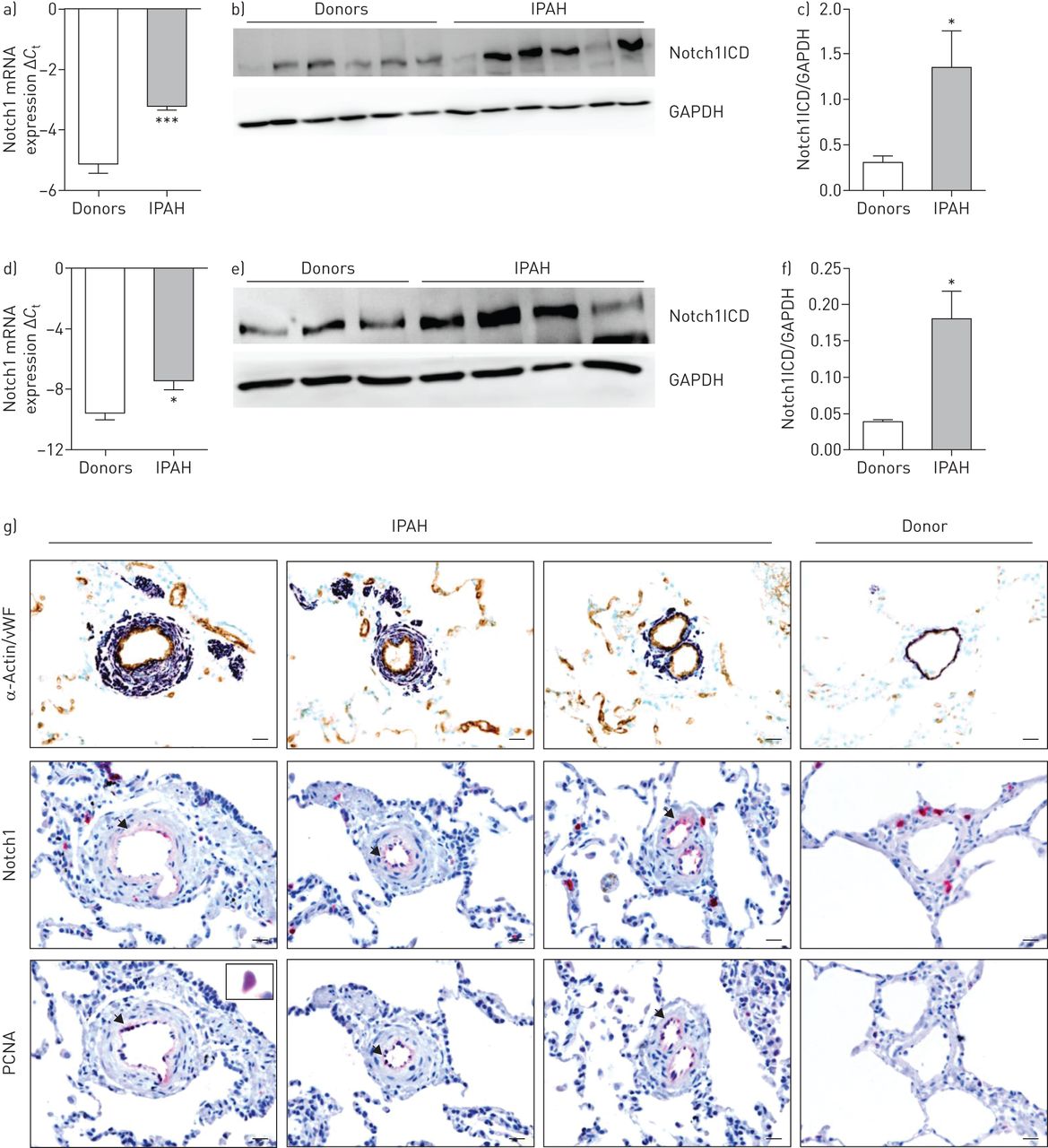
mRNA expression of Notch1 and protein expression of Notch1ICD was strongly upregulated in lung homogenates from patients with IPAH compared with donors (figure 1a–c). Further Notch1 expression was assessed in microdissected pulmonary vessels and plexiform lesions (figure 1d and online supplementary figure S2). There was a significant increase in mRNA expression of Notch1 observed in microdissected vessels from IPAH patients compared with donor tissues, but not in plexiform lesions. Similarly, at the protein level, we observed a substantial increase in Notch1ICD expression in IPAH pulmonary arteries (figure 1e and f). A strong immunoreactivity of Notch1 was observed in the intimal layer of pulmonary arteries of IPAH lungs (figure 1g), which clearly colocalised with proliferating cell nuclear antigen (PCNA). Significant changes, at both the mRNA and protein level, were not observed for other Notch receptors and ligands in IPAH lung homogenates except for DLL1 ligand (online supplementary figure S1).

Notch1 expression and localisation in human donor and idiopathic pulmonary arterial hypertension (IPAH) lungs. mRNA expression of Notch1 in a) lung homogenates and d) pulmonary arteries of donors and IPAH patients. Regulation at the mRNA level was analysed by real-time PCR using the cycle threshold (Ct) method (n=7). Western blot analysis of Notch1 intracellular domain (Notch1ICD) in b) lung homogenates and e) pulmonary arteries of donors and IPAH patients, followed by c, f) densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was taken as the loading control. *: p<0.05; ***: p<0.001 versus donors. g) Representative immunostaining micrographs of human lung sections from donors and IPAH patients. Staining was undertaken for Notch1, proliferating cell nuclear antigen (PCNA), von Willebrand factor (vWF; brown; endothelial cells) and α-actin (purple; smooth muscle cells). Scale bar: 20 μm.
Expression of Notch1 in hPAECs under hypoxia and its effect on hypoxia-induced proliferation
Exposure of hPAECs to hypoxia (1% O2) resulted in upregulation of Notch1 mRNA expression (figure 2a) and Notch1ICD protein expression (figure 2b and c). Based on the presence of hypoxia response element sites in the Notch1 promoter (online supplementary figure S3), a dual luciferase assay was performed in HEK cells with the pGL4-Notch1 promoter construct. Luciferase activity was significantly increased after hypoxia exposure compared with normoxia (figure 2d). More interestingly, hypoxia-induced hPAECs proliferation was decreased by Notch1 knockdown compared with si-Scr-transfected cells (figure 2e), indicating a central role of Notch1 in the proliferative phenotype of hPAECs under hypoxia. Specific knockdown of Notch1ICD (85% knockdown efficiency) along with decreased mRNA expression of the Notch target Hes1 gene was achieved by Notch1 siRNA with no effect on Notch2ICD and Notch4ICD (figure 3b and c, and online supplementary figure S4A–C).

Expression of Notch1 in human pulmonary arterial endothelial cell (hPAECs) under hypoxia and its effect on hypoxia-induced proliferation. hPAECs were exposed to hypoxia (1% O2; HOX) and normoxia (NOX) for 24 h. a) mRNA expression of vascular endothelial growth factor (VEGF) and Notch1 in hPAECs exposed to HOX. Regulation at the mRNA level was analysed by real-time PCR using the cycle threshold (Ct) method. *: p<0.05; ***: p<0.001 versus NOX control. b) Western blot analysis of Notch1 intracellular domain (Notch1ICD) and hypoxia-inducible factor (HIF)-1α followed by c) densitometric analysis. β-Actin was taken as the loading control. *: p<0.05 versus NOX. d) Human embryonic kidney cells were transfected with human Notch1 promoter/reporter constructs and exposed to hypoxia for 24 h, and relative luciferase activity in cell extracts was measured with a luminometer and expressed as relative luciferase activity. All values represent mean±sem (n=9). **: p<0.01 versus NOX. e) hPAEC proliferation as measured by bromodeoxyuridine (BrdU) incorporation 48 h after small interfering RNA (siRNA) targeting Notch1 mRNA (si-Notch1) followed by hypoxia exposure. ***: p<0.001 versus scrambled siRNA (si-Scr) (HOX).

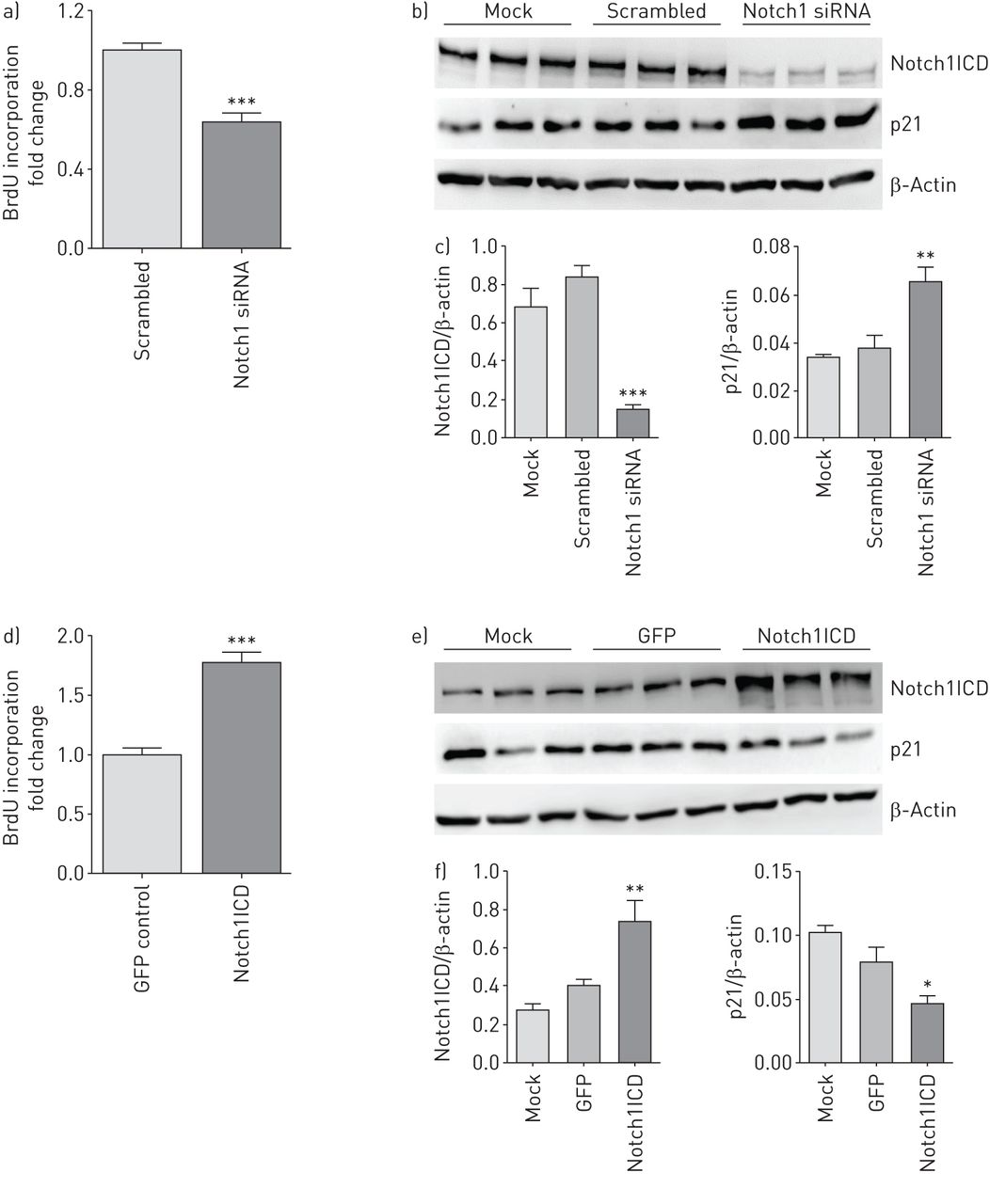
Effect of Notch1 knockdown and overexpression on growth medium-induced proliferation and cell cycle regulators in human pulmonary arterial endothelial cell (hPAECs). hPAECs were transiently transfected with small interfering RNA (siRNA) targeting Notch1 mRNA (si-Notch1) or Notch1 intracellular domain (Notch1ICD) plasmid for 48 h. hPAECs treated with transfection reagent alone is the mock. hPAEC proliferation induced by growth medium as measured by bromodeoxyuridine (BrdU) incorporation 48 h after a) si-Notch1 and d) Notch1ICD plasmid transfection. ***: p<0.001 versus scrambled siRNA (si-Scr) or green fluorescent protein (GFP) control (n=5). Effect of b) si-Notch1 and e) Notch1ICD overexpression on cell cycle regulators as analysed by Western blotting with antibodies against p21, followed by c, f) densitometric analysis of the blots. β-Actin was taken as the loading control. *: p<0.05; **: p<0.01; ***: p<0.001 versus mock. n=3 in each group.
Effect of Notch1 knockdown and overexpression on proliferation of hPAECs
In vitro studies were performed using Notch1 siRNA (si-Notch1) and Notch1ICD plasmid to investigate the functional role of Notch1 in hPAECs. Importantly, Notch1 knockdown decreased growth medium-induced proliferation (∼40%) compared with si-Scr-transfected hPAECs (figure 3a). Moreover Notch1 knockdown increased expression of p21, an endogenous cell cycle inhibitor (figure 3b and c), suggesting Notch1 is involved in cell cycle regulation.
Functional overexpression of Notch1ICD, confirmed by a significant increase in mRNA expression of Hes1 (online supplementary figure S5B), led to a substantial increase in growth medium-stimulated proliferation of hPAECs (77%) compared with empty vector-transfected cells (figure 3d) and a concomitant decrease in expression of p21 (figure 3e and f). In addition to endothelial cells, Notch1 is reportedly expressed in vascular smooth muscle cells [14, 21]; however, there was no change observed in growth medium-stimulated proliferation of human PASMCs (hPASMCs) after knockdown or constitutive expression of Notch1 (online supplementary figure S6A and F).
Effect of Notch1 knockdown and overexpression on apoptosis of hPAECs
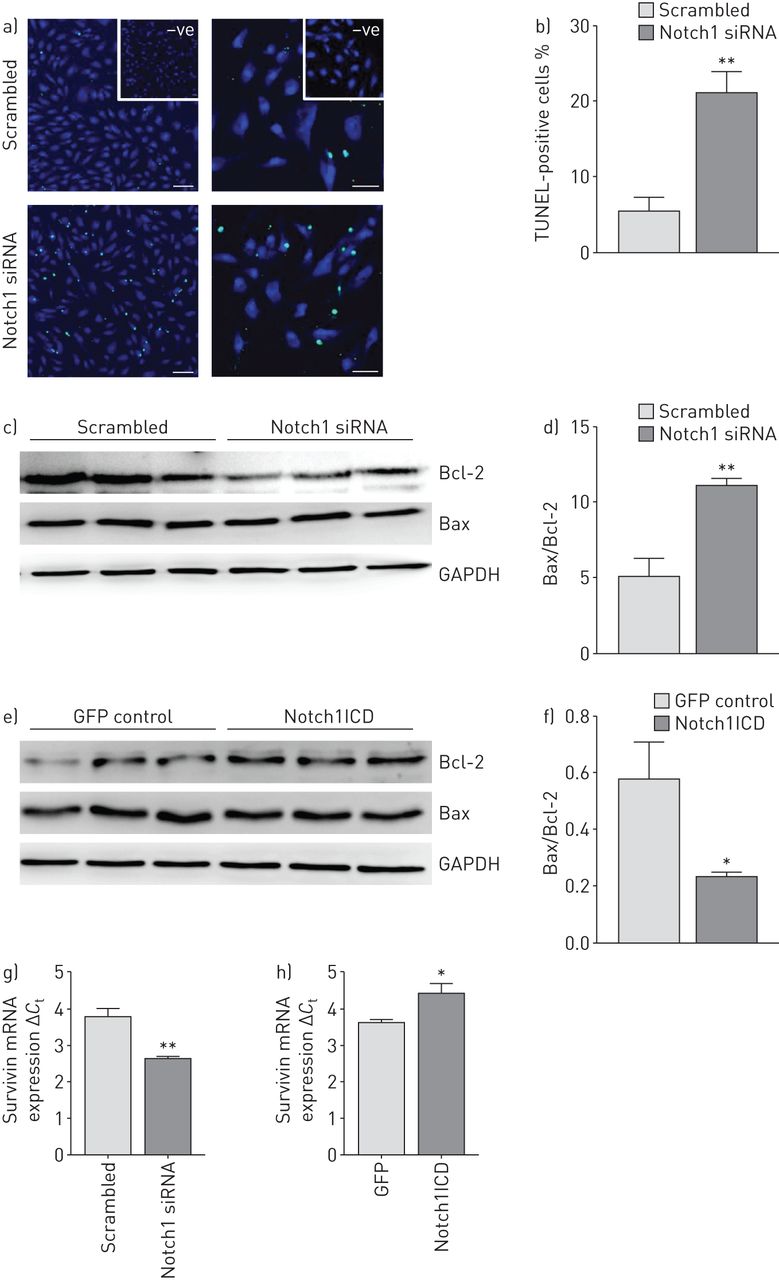
Knockdown of Notch1 led to ∼20% increase in the number of apoptotic cells compared with si-Scr-transfected cells measured by TUNEL (terminal deoxynucleotidyl transferase dUTP nick end-labelling) assay (figure 4a and b). Additionally, Western blot analysis demonstrated a significant increase of the Bax/Bcl-2 ratio in si-Notch1-transfected cells (figure 4c and d), indicative of increased apoptosis. Transfection of hPAECs with the Notch1ICD expression plasmid led to a significant decrease in the Bax/Bcl-2 ratio (figure 4e and f) compared with empty vector. mRNA expression of Survivin was decreased by Notch1 knockdown while vice versa it was observed with Notch1ICD overexpression (figure 4g and h).
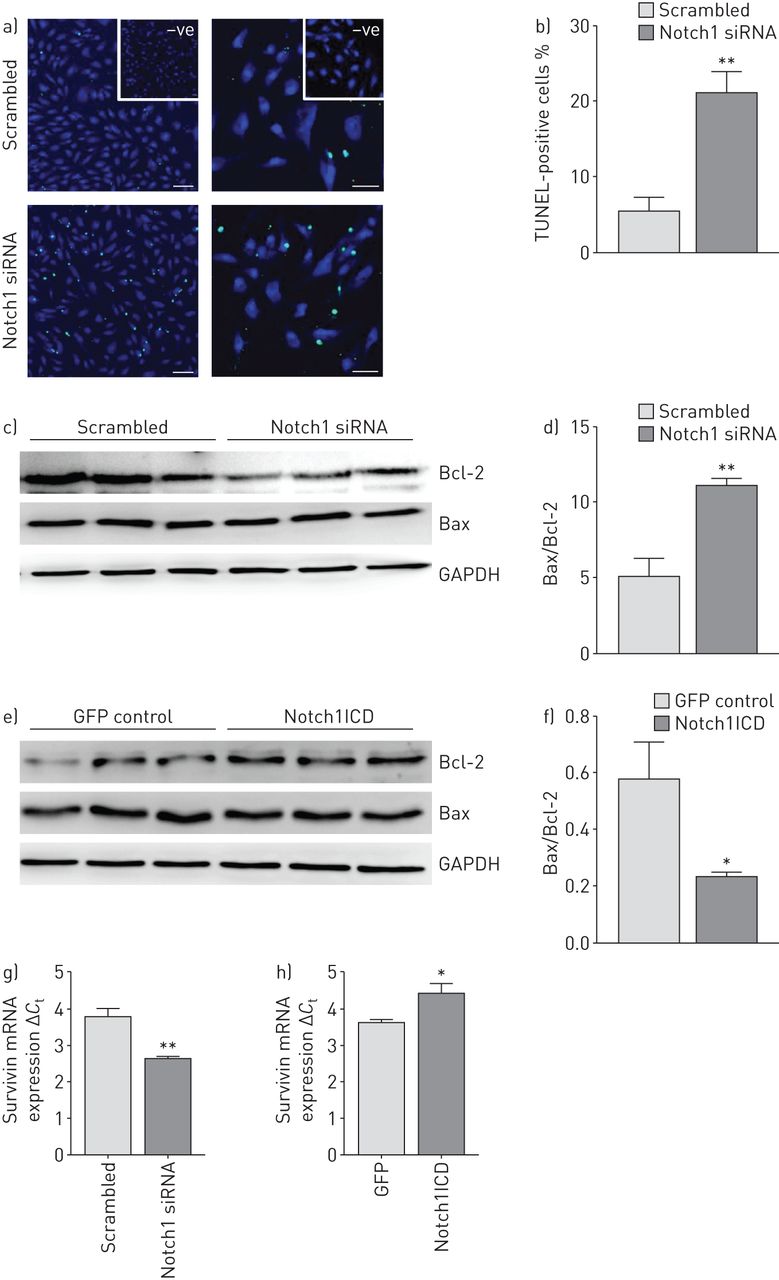
Effect of Notch1 knockdown and overexpression on apoptosis of human pulmonary arterial endothelial cell (hPAECs). a) Representative images of scrambled small interfering RNA (siRNA) (si-Scr)- and siRNA targeting Notch1 mRNA (si-Notch1)-transfected cells that were assessed for apoptosis using an in situ cell death detection method (TUNEL (terminal deoxynucleotidyl transferase dUTP nick end-labelling) assay) 48 h after transfection. Scale bar: 50 μm. b) Percentage of apoptotic (TUNEL-positive) cells as counted using STEPanizer software. Effect of c) Notch1 knockdown and e) Notch1 intracellular domain (Notch1ICD) overexpression was analysed on apoptotic proteins by Western blotting with antibodies against Bcl-2 and Bax followed by d, f) densitometric analysis of the blots. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was taken as the loading control. *: p<0.05; **: p<0.01 versus b, c) si-Scr or d) green fluorescent protein (GFP) control. n=3 in each group. mRNA expression of apoptotic protein Survivin as measured by real-time PCR in g) si-Notch1- or h) Notch1ICD-transfected cells. *: p<0.05; **: p<0.01 versus si-Scr or GFP control.
Effect of pharmacological inhibition of Notch signalling on proliferation, migration and angiogenic response of hPAECs
Treatment with dibenzazepine (DBZ; a γ-secretase inhibitor) decreased γ-secretase activity by 50% after 8 h in hPAECs as observed by the reduced protein levels of Notch1ICD (figure 5a and b) and decreased mRNA expression of Hes1 (figure 5c). 5% Fetal calf serum (FCS) resulting in a marked increase in proliferation of hPAECs (by 88%), which was reduced to less than half (up to 66%) by 20 μM DBZ (figure 5d). Interestingly, proliferation induced by VEGF, a potent mitogen for hPAECs, was also reduced effectively by 20 μM DBZ by 56% (figure 5e). Furthermore, FCS-induced migration of hPAECs was significantly reduced by DBZ in a dose-dependent manner with 20 μM DBZ showing a reduction by 50% (figure 5f and g), although no effect of DBZ was observed on the tube formation response of the hPAECs (online supplementary figure S7). Data from work dissecting Notch3 signalling in PAH revealed that DAPT (N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester), another γ-secretase inhibitor, was able to reduce proliferation of hPASMCs [11]. Similarly, DBZ significantly decreased DNA synthesis in both 5% FCS- and platelet-derived growth factor (PDGF)-BB-stimulated hPASMCs in a dose-dependent manner (online supplementary figure S8a and b).

Effect of pharmacological inhibition of Notch signalling on serum or vascular endothelial growth factor (VEGF)-induced proliferation and migration of human pulmonary arterial endothelial cell (hPAECs). a) Western blot analysis of Notch1 intracellular domain (Notch1ICD) after treatment of hPAECs with dibenzazepine (DBZ) with glyceraldehyde 3-phosphate dehydrogenase (GAPDH) as the loading control. b) Quantification of the Western blot. GAPDH was taken as the loading control. **: p<0.01; ***: p<0.001 versus 0 h. c) mRNA expression of Hes1 in hPAECs after 24 h treatment with DBZ as analysed by real-time PCR. All values are expressed as ΔCt. ***: p<0.001 versus dimethylsuphoxide (DMSO) control. hPAECs were serum starved and stimulated with d) 5% fetal calf serum (FCS) or e) 10 ng·mL−1 VEGF in the presence of various concentrations of DBZ or DMSO (–) and measured for changes in proliferation by bromodeoxyuridine (BrdU) incorporation. All data are expressed as fold increase of the nonstimulated group and represent mean±sem (n=5). f) hPAECs were serum starved for 12 h and treated with 10 M mitomycin C for 2 h. Cells were then seeded on transwell inserts and lower chambers were filled with 5% FCS in the presence of various concentrations of DBZ or DMSO (–). After 16 h, cell migration was measured by counting the crystal violet-positive cells under a microscope. *: p<0.05; ***: p<0.001 versus nonstimulated; #: p<0.05 versus d, f) FCS- or e) VEGF-treated group. g) Representative images from the transwell migration assay. Scale bar: 100 μm.
Notch1 expression in SUHx rats and therapeutic assessment of a γ-secretase inhibitor (AMG2008827) in SUHx rats
Previous reports suggested that rats exposed to hypoxia after injection of the VEGFR-2 inhibitor SU5416 exhibit neointimal lesions, characterised by hyperproliferative and apoptosis-resistant endothelial cells [22]. We observed a strong upregulation of Notch1 at the mRNA and protein level in lungs of SUHx rats compared with healthy rats (figure 6a–c), concomitant with our findings in IPAH lungs. Also, Notch3 expression was found to be strongly upregulated (online supplementary figure S9). To assess the therapeutic potential of Notch inhibition in SUHx pulmonary hypertension, rats were treated 3 weeks after Sugen injection and hypoxia exposure with a γ-secretase inhibitor (GSI; AMG2008827; Amgen, Thousand Oaks, CA, USA), which inhibits Notch1 signalling in a cellular reporter assay with an IC50 (the half maximal inhibitory concentration) of 1.28 nM (online supplementary figure S10), or placebo for 14 days. GSI treatment of SUHx rats resulted in a remarkable decrease in right ventricular systolic pressure (figure 6d) and increase in cardiac index (figure 6e) as compared with placebo-treated SUHx rats. Additionally, GSI treatment significantly increased the pulmonary artery acceleration time/pulmonary arterial ejection time ratio and tricuspid annular plane systolic excursion (figure 6f and g), and decreased the total pulmonary vascular resistance index and right ventricular internal diameter (figure 6h and i) as measured by echocardiography. With respect to vascular remodelling, GSI treatment led to a reduction in the neointima/media ratio and medial wall thickness (online supplementary figure S11).

Notch1 expression and localisation in lungs from hypoxia/SU5416 (SUHx)-treated rats and effect of a γ-secretase inhibitor (GSI; AMG2008827) on SUHx-induced pulmonary hypertension in rats. a) mRNA expression of Notch1 in lung homogenates of healthy and SUHx rats. Regulation at the mRNA level was analysed by real-time PCR using the cycle threshold (Ct) method (n=4). b) Western blot analysis of Notch1 intracellular domain (Notch1ICD) in lung homogenates of healthy and SUHx rats, followed by c) densitometric analysis. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was taken as the loading control. ***: p<0.001 versus healthy. d–i) Influence of GSI on the haemodynamics and right heart function in SUHx rats. BW: body weight. Echocardiography followed by physiological measurements was carried out on GSI-treated (3 mg·kg BW−1)/placebo-treated SUHx rats and healthy rats 35 days after initiation of SUHx treatment. d) Right ventricular systolic pressure (RVSP) as measured by right heart catheterisation, and e) cardiac index, f) pulmonary arterial acceleration time (PAAT)/pulmonary arterial ejection time (PAET), g) tricuspid annular plane systolic excursion (TAPSE), h) right ventricular internal diameter (RVID) and i) total pulmonary vascular resistance index (TPVRi) of the rats as measured by echocardiography. n=5 healthy, n=7 SUHx placebo, n=5 GSI-treated SUHx. *: p<0.05; **: p<0.01; ***: p<0.001 versus SUHx+placebo.
Discussion
Our investigation of the role of Notch signalling in PAH led us to the following key observations. 1) Notch1 expression and cleavage is increased in lungs and pulmonary arteries of IPAH patients and in lungs of SUHx rats. 2) Notch1 positively regulates proliferation of hPAECs via p21, and negatively regulates apoptosis via Bcl-2 and Survivin, with no effect on proliferation of hPASMCs, supporting endothelial-specific effects of Notch1. 3) Hypoxia is one of the stimuli leading to increased Notch1 signalling in hPAECs. 4) Inhibition of Notch signalling in vitro by DBZ effectively attenuated proliferation in both hPAECs and hPASMCs, and in vivo administration of GSI (AMG2008827) potently improved right ventricular systolic pressure and right heart function of SUHx rats.
Endothelial cells in pulmonary arteries of PAH patients exhibit a hyperproliferative and anti-apoptotic phenotype [5, 23]. Somatic mutations in bone morphogenetic protein receptor type II (BMPRII) and Bax genes have been identified in patients with IPAH [6, 24], which may provide a growth advantage to altered endothelial cells. However, BMPRII mutations are observed only in 70% of familial PAH and not all intimal lesions harbour somatic mutations. Hence, other molecular players need to be investigated. Based on our present study, it is compelling to speculate that Notch1 could qualify as one such molecular mediator and that modulation of Notch1 signalling in the endothelium may be beneficial for patients with PAH.
An initial hint about the relevance of Notch1 signalling in PAH was provided by our screening studies which showed an increased expression and activation of Notch1 signalling in the pulmonary vasculature of IPAH patients. Additionally, we observed increased Notch1 expression in endothelial cells in IPAH vessels compared with donor vessels. This change was correlated with increased PCNA staining, suggesting that Notch1 could be involved in endothelial proliferation and neointima formation. To the best of our knowledge, this is the first study demonstrating increased Notch1 activation in the pulmonary vasculature of IPAH patients. SUHx rats, reported to mirror the hyperproliferative endothelial cell aetiology of severe human PAH [22, 25], exhibited increased Notch1 expression and cleavage in lungs compared with healthy rats, further suggesting a role of Notch1 in endothelial proliferation and survival in vivo. Activation of Notch receptor is dependent on the interaction with ligands displayed on adjacent cells [10]. Increased expression of the DLL1 ligand in IPAH patients could represent one possible way of inducing Notch1 activation. However, certain reports have suggested ligand-independent activation of Notch signalling. An in vitro study has demonstrated increased cleavage and activation of Notch1 in embryonic stem cell-derived VEGFR-2+ cells under shear stress [26]. Hyperproliferative altered endothelial cells generally observed at branching points of pulmonary arteries in IPAH patients are believed to be an outcome of increased shear stress [23] and SUHx treatment in rats has also been shown to mirror these shear stress conditions [22], implicating shear stress as a direct Notch1 activator. Furthermore, additional PAH contributing factors such as reactive oxygen species or HIF-1α activation have also been linked to Notch1 activation. In order to define the mechanisms involved in Notch1 activation, we explored hypoxia as an initial stimulus. Hypoxia has been described as an important trigger leading to remodelling of vessels in pulmonary hypertension [27]. In our studies, Notch1 expression, cleavage and promoter activity were strongly upregulated in hypoxia-exposed hPAECs and HEK cells (dual luciferase assay). This increase may be attributed to HIF-1α, as HIF-1α-binding sites are present in the Notch1 promoter sequence. Interestingly, elevated levels of HIF-1α have been detected in arterial lesions in IPAH lungs and in IPAH PAECs [28]. A recent study has further revealed activation of Notch1 in cancer cell lines under hypoxia via HIF-1α, contributing to increased proliferation, migration and survival of the cells [29]. Notch1 inhibition reversed hypoxia-induced proliferation in hPAECs. Hence, we conclude that hypoxia potentiated Notch1 signalling in hPAECs, which in turn contributed to increased proliferation.
Similarities between cancerous cells and altered endothelial cells in PAH patients [3, 30], and the established role of Notch1 as an oncogene [31, 32], led us to examine its contribution in endothelial proliferation and apoptosis. In our studies, Notch1 downregulation resulted in ∼40% decrease in proliferation of hPAECs, while Notch1ICD overexpression led to a significant ∼75% increase in proliferation. Earlier reports implicated Notch1 in the regulation of endothelial proliferation, albeit with conflicting conclusions. In human aortic endothelial cells, Notch1ICD overexpression was shown to inhibit proliferation of the cells via repression of mitogen-activated protein kinase and phosphoinositide 3-kinase/Akt pathway [16] and p21 [17]. Contradicting these reports, Takeshita et al. [18] demonstrated that Notch1 is essential for VEGF-induced proliferation, migration and survival of endothelial cells. Another study linked reduced Notch1 cleavage to decreased PCNA levels in endothelial cells, when cocultured with Jagged1-deficient vascular smooth muscle cells [19]. To explore the mechanism involved, expression of cell cycle regulatory proteins was examined. We observed a marked increase in protein levels of the cell cycle inhibitor p21 upon Notch1 knockdown, while Notch1ICD overexpression was associated with decreased expression of p21. p21, a well-known cyclin-dependent kinase inhibitor, has been described as a direct target of RBP-Jκ-dependent Notch1 signalling [17, 33], where the authors demonstrated repression of p21 promoter activity under Notch1 activation in human umbilical vein endothelial cells. Our results support a positive role of Notch1 in the proliferation of hPAECs via an effect on p21 expression. Interestingly, siRNA-mediated Notch1 knockdown or Notch1ICD overexpression did not show any effect on proliferation of hPASMCs, indicating that other Notch homologs are more important in regulating the proliferative potential of PASMCs. Indeed, Notch3 has been described to be essential for aberrant proliferation of PASMCs in IPAH patients [11]. This finding further supports our hypothesis of endothelial-specific effects of Notch1.
Resistance to apoptosis is another important characteristic of altered endothelial cells found in patients with PAH [5, 6]. Notch1 knockdown led to a small but significant (20%) increase in the number of TUNEL-positive apoptotic endothelial cells. Many reports have proposed and further demonstrated that the Bcl-2/Bax ratio might govern the sensitivity of cells to apoptotic stimuli [34]. Interestingly, we were able to observe a significant decrease of the Bax/Bcl-2 ratio, indicative of increased apoptosis upon treatment of hPAECs with si-Notch1. In agreement, constitutive expression of Notch1 ICD led to an increase in the Bax/Bcl-2 ratio. Survivin, a member of the mammalian “inhibitor of apoptosis” family, is shown to be expressed in cancers and remodelled pulmonary arteries of PAH patients [35, 36]. We observed a positive effect of Notch1 on Survivin mRNA expression, indicating Survivin is a downstream target of Notch1 signalling, in agreement with various studies suggesting Notch1 is an upstream regulator of Survivin in cancer [37]. Previous studies have defined an anti-apoptotic role of Notch1 signalling in both animal and in vitro cell models [38], in the field of development and cancer. However, not much is known about the involvement of Notch1 in endothelial apoptosis. Our results suggest a role of Notch1 in promoting survival of hPAECs by regulating expression of Bcl-2 and Survivin.
Pharmacological inhibition of Notch signalling achieved by γ-secretase inhibitors has been extensively used in various studies utilising cell lines and animal models for different cancers [39, 40] to establish the potential utility of γ-secretase inhibitor-based treatments. In order to examine the relevance of γ-secretase inhibitors as a therapy in experimental pulmonary hypertension, we used DBZ for our in vitro studies. DBZ was able to significantly reduce serum and VEGF-induced proliferation of endothelial cells. In fact, cross-talk between VEGF and Notch signalling has already been reported. VEGF was shown to increase expression of Notch receptors and ligands in endothelial cells [41]. Additionally, VEGF-induced postnatal angiogenesis involving endothelial proliferation and migration is mediated via γ-secretase and Notch1 activation [18]. VEGF plasma levels are elevated in patients with severe PAH, and VEGF as well as VEGFR-2 are strongly expressed in plexiform lesions [4]. DBZ further exhibited a strong inhibitory effect on migration of hPAECs stimulated by FCS, although there was no significant effect on the angiogenic response. It has been reported that IPAH PAECs show increased migratory potential compared with control cells. However, no greater angiogenic potential of IPAH PAECs was observed, but rather a less orderly tube formation with thinner branches. Pulmonary angiogenesis during development of PAH may act as a protective mechanism against loss of the pulmonary microvasculature due to endothelial injury and dysfunction, although severe PAH is characterised by deregulated angiogenesis where endothelial cell proliferative lesions consist of disorganised, alternating areas with a solid core of cells and other areas with variable stages of capillary/blood vessel formation [42]. The substantial effects of Notch modulation observed on the proliferation, migration and apoptosis of hPAECs, but not angiogenesis, hint strongly towards a beneficial role of Notch inhibition for treating PAH without having any negative effect on pulmonary angiogenesis. In line with previous findings [11], serum and PDGF-induced proliferation of hPASMCs was effectively reduced by DBZ. The effect of DBZ on PDGF-BB induced proliferation could be either due to a direct effect of Notch on cell proliferation markers or due to cross-talk with PDGF signalling. Indeed, Notch3 is shown to regulate PDGF receptor β expression in vascular smooth muscle cells [43].
Importantly, the therapeutic potential of GSI (AMG2008827; 3 mg·kg−1) in vivo on several disease parameters of established SUHx-induced pulmonary hypertension in rats was assessed. The inhibitor markedly improved the haemodynamic parameters (right ventricular systolic pressure and cardiac index) and right heart function in SUHx rats compared with placebo-treated rats. Furthermore, reduction in neointima formation and medial wall thickness with GSI treatment were also observed. However, a higher dose of the inhibitor or prolonged administration may be needed to show further sustained effects. Taken together, these data indicate a protective role of Notch inhibition in experimental PAH.
Limitations of the study
The present study elucidates a clear mechanism of regulation of endothelial proliferation and survival by Notch1 signalling in vitro and in vivo. Further studies are needed to dissect the molecular mechanism (ligand dependent and independent) leading to Notch1 activation in the disease state and to assess the efficacy of GSI treatment in combination with other compounds that have been previously reported to reduce pulmonary hypertension in the SUHx model.
Conclusion
These data describe a novel pathway involving Notch1 signalling, which regulates a pro-proliferative and pro-survival phenotype in PAECs (figure 7). This pathway appears to be further activated by hypoxia stimulation. Both in vitro and in vivo data using pharmacological γ-secretase inhibitors suggest that Notch1 is a potential therapeutic target in PAH.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
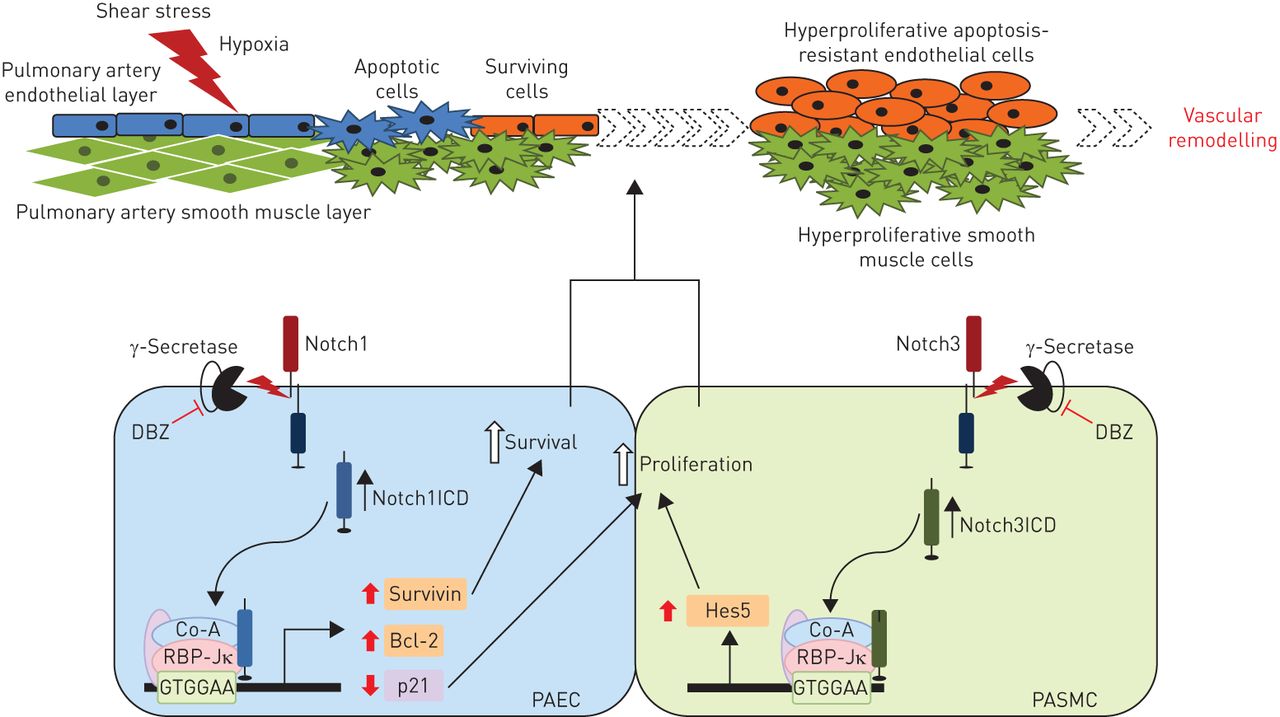
Notch signalling in pulmonary arterial hypertension (PAH). Schematic representation of the proposed role of Notch signalling in pulmonary vascular remodelling in PAH. Notch1ICD: Notch1 intracellular domain; DBZ: dibenzazepine; Co-A: coactivator; RBP-Jκ: recombination signal sequence binding protein Jκ; PAEC: pulmonary artery endothelial cell; PASMC: pulmonary artery smooth muscle cell; Hes5: transcription factor Hes5.
Acknowledgements
We acknowledge Ewa Bieniek and Christina Vroom (Universities of Giessen and Marburg Lung Center, Giessen, Germany), and Yanina Knepper (Max Planck Institute for Heart and Lung Research, Bad Nauheim, Germany) for their valuable technical assistance.
Footnotes
This article has supplementary material available from erj.ersjournals.com
Support statement: This work was supported by funding from the Universities of Giessen and Marburg Lung Center (UGMLC) and Max Planck (MPI) Society. Funding information for this article has been deposited with FundRef.
Conflict of interest: None declared.
- Received May 18, 2015.
- Accepted May 6, 2016.
- Copyright ©ERS 2016
References










