





Abstract
This review is devoted to the distinct associations of inflammatory bowel diseases (IBD) and chronic liver disorders with chronic airway diseases, namely chronic obstructive pulmonary disease and bronchial asthma, and other chronic respiratory disorders in the adult population. While there is strong evidence for the association of chronic airway diseases with IBD, the data are much weaker for the interplay between lung and liver multimorbidities. The association of IBD, encompassing Crohn's disease and ulcerative colitis, with pulmonary disorders is underlined by their heterogeneous respiratory manifestations and impact on chronic airway diseases. The potential relationship between the two most prevalent liver-induced pulmonary vascular entities, i.e. portopulmonary hypertension and hepatopulmonary syndrome, and also between liver disease and other chronic respiratory diseases is also approached. Abnormal lung function tests in liver diseases are described and the role of increased serum bilirubin levels on chronic respiratory problems are considered.
Abstract
Lung–gut cross-talk: the association of IBD with respiratory multimorbitidies, including COPD and bronchial asthma http://ow.ly/UT94a
Introduction
This review addresses the “cross-talk” between a spectrum of chronic respiratory disorders and inflammatory bowel diseases (IBD), i.e. Crohn's disease and ulcerative colitis, as well as the connections between pulmonary vascular syndromes, namely portopulmonary hypertension (POPH) and hepatopulmonary syndrome (HPS), induced by chronic hepatic diseases, predominantly liver cirrhosis. After addressing the most salient epidemiological and clinical characteristics of lung, intestinal and liver disorders, we approach pathophysiological and pathobiological aspects to assist in diagnosing and managing these complex and challenging comorbidities. The connections of the lungs with the gut and the liver are heterogeneous and complex, with several unknowns from a mechanistic viewpoint.
The evidence of gastrointestinal and liver multimorbidities that can coexist in adult patients with chronic respiratory diseases, in particular chronic airway diseases, is relatively limited and remains insufficiently investigated. The following four publications are good examples of this statement. A longitudinal study about the spectrum and prevalence of multimorbidities in a population of >1600 patients with chronic obstructive pulmonary disease (COPD) over 4 years reported both gastro-oesophageal reflux disease (GORD) (18% and 20% in survivors and nonsurvivors, respectively) and liver cirrhosis (<2% each) alone [1]. A study of ∼20 000 patients aimed at assessing the impact of chronic conditions on tertiary hospital admissions revealed that the prevalence of liver cirrhosis in the COPD population was <5%, a figure close to the prevalence of COPD observed in the hepatic cirrhotic cohort [2]. A third, more modest, study of 215 patients admitted to hospital (28% diagnosed with COPD) observed no differences in the number of multimorbidities between patients with and without COPD. However, gastrointestinal, haematological and uro-nephrological disorders were more prevalent in patients with COPD and their hospital stay was longer [3]. Finally, a landmark study of the role played by multimorbidities in systemic inflammation in 213 patients with COPD, after excluding active inflammatory disease, did not make any reference to IBD or hepatic diseases [4]. Alternatively, in a review of the co-occurrence of multimorbidities in bronchial asthma, gastrointestinal or liver diseases, other than the common coexistence of GORD [5], were not reported at all.
It is anticipated that studies of the prevalence and incidence of multimorbidities affecting chronic respiratory conditions and IBD have been more extensively investigated than those directed at the association with liver disorders. The latter are fewer and scant, so that the design of appropriate epidemiological studies in this field seems to be warranted.
Inflammatory bowel diseases
IBD is a term that describes a miscellany of inflammatory diseases of the gastrointestinal tract, namely Crohn's disease and ulcerative colitis, the two most common entities comprising IBD [6]. However, from a pathophysiological viewpoint the two conditions are distinct. Crohn's disease is a relapsing inflammatory disease, mainly affecting any site of the intestinal tract manifesting with transmural, sporadic digestive tract lesions [7]. By contrast, ulcerative colitis is a type of colitis that causes inflammation affecting the colon and rectum only, characterised by continuous, superficial ulceration of the colon and leaving the rest of the gastrointestinal unscathed [8]. Unlike Crohn's disease, with a tendency to recur following surgery, surgical removal of the colon and rectum resolves ulcerative colitis. Clinically, both conditions share excessive daily bowel movements, severe abdominal pain, diarrhoea, weight loss, malnutrition and intestinal bleeding. It is calculated that ∼25% of patients with IBD have evidence of extra-intestinal, systemic manifestations, including respiratory conditions [6, 9–11]. Although the causes of IBD remain unclear, several factors, such as genes, environment, the gut microbiome and inflammatory dysfunction, have been invoked [8, 12].
Respiratory manifestations of inflammatory bowel diseases
Both acute and chronic respiratory manifestations of IBD have been reported for many years, but appear less often than manifestations in other organs [13], at variance with the contention that the lung and gut share a common embryologic origin and thus might have similar susceptibility to immunologically mediated multimorbidities. Pulmonary complications occur more commonly with ulcerative colitis than with Crohn's disease. Respiratory involvement may occur at any age and chronic airway diseases appear more often in females than in males [13]. Although respiratory symptoms commonly develop after the diagnosis of IBD has been made, they may occasionally precede, or be concomitant with those of IBD [11].
Most respiratory complications involve the airways at any level and may include a wide spectrum of epiglottitis, tracheal stenosis, bronchiectasis, chronic bronchitis, COPD, bronchial asthma and/or bronchiolitis [14]. From an endoscopic viewpoint, there can be intense erythema, mucosal swelling and deformation of the airway lumen. Biopsy studies have revealed mucosal ulceration, thickening of the basement membrane and marked infiltration by neutrophils and plasma cells. Another major category of pulmonary complications of IBD is interstitial lung diseases, such as cryptogenic organising pneumonia, sarcoidosis, interstitial fibrosis and pulmonary infiltrations with peripheral blood and sputum eosinophilia [15]. Medications, such as mesalazine, sulfasalazine and methotrexate can be the most frequent cause of respiratory manifestations of IBD [16] and must always be considered in the differential diagnosis of new-onset interstitial lung disease. Increased risk of developing tuberculosis infection in patients with IBD during tumour necrosis factor-α inhibitor therapy should be also taken into consideration [17].
Patients with IBD, often clinically asymptomatic and with normal chest radiographs may exhibit a variety of lung function abnormalities, including obstructive and restrictive spirometric defects. An abnormal decreased transfer factor (diffusing capacity) of the lung for carbon monoxide (TLCO) has been also detected in active IBD [18, 19], suggesting subclinical interstitial pulmonary involvement. Under these circumstances, examination using bronchoalveolar lavage in patients with Crohn's disease without clinical evidence of pulmonary involvement revealed the presence of a lymphocytic alveolitis, with predominant CD4+ T-lymphocytes [20, 21].
In most patients with respiratory disorders associated with IBD, glucocorticosteroids given by inhalation or systemically usually result in a rapid control of symptoms and lung function [11, 22, 23], especially in patients without associated severe chronic airway diseases. The phosphodiesterase (PDE)4 inhibitor roflumilast, recommended for the treatment of severe to very severe COPD patients with associated mucus hypersecretion and frequent exacerbations has shown potential efficacy in animal studies of IBD [24, 25]. PDE4 inhibitors affect IBD in different ways including anti-inflammatory, antidepressant and antifibrotic effects. Tetomilast, a second-generation PDE4 inhibitor, showed promising effects in phase II studies, with a better safety profile [26, 27].
Impact of IBD in chronic airway diseases
In common with many other extraintestinal manifestations of IBD, the activity of the intestinal disease is not thought to be linked to the development and progression of IBD-associated chronic airway diseases. However, there is a four-fold increased prevalence of IBD in a cohort of patients with chronic airway diseases, including chronic bronchitis and cough, bronchiectasis, COPD and asthma [28] (table 1), akin to the predominance of nonasthmatic airway disease with productive cough observed in the majority of clinical series and patients with ulcerative colitis [11, 13, 22, 29–31]. Moreover, in a large population-based study of ∼180 000 patients with COPD aimed at investigating the occurrence of IBD, the risk of IBD in COPD patients was higher than that related to smoking alone [32]. A more recent study of >200 000 females looking at the associations between smoking and IBD before the diagnosis of incident cases of Crohn's disease and ulcerative colitis concluded that compared to never-smokers current smoking was associated with an increased risk of Crohn's disease, but not of ulcerative colitis [33]. Compared to the general population, it has been observed that over a 5-year period, the average incidence of Crohn's disease is increased in patients with asthma and COPD [34] (figure 1); the incidences of Crohn's disease in asthma and COPD were 27% and 55%, respectively, while that of ulcerative colitis was 30% higher in patients with COPD.

Age-specific incidence rates of Crohn's disease and ulcerative colitis in patients with a) asthma and b) chronic obstructive pulmonary disease (COPD). The highest incidence for Crohn's disease (34.3 cases per 100 000 person-years) was in the 20–29-year-old age subset and that for ulcerative colitis (14.9 cases per 100 000 person-years) was in the 30–39-year-old range, in the asthma cohort. By contrast, the highest incidence of Crohn's disease (35.5 cases per 100 000 person-years) occurred in the 50–59-year-old age subset and that for ulcerative colitis (24.9 cases per 100 000 person-years) was in the 60–69-year-old group in the COPD population. Reproduced from [34] with permission from the publisher.
Incidence or prevalence ratios of Crohn's disease and ulcerative colitis in patients with chronic respiratory diseases
Mechanisms of lung–gut interplay
An impairment of intestinal permeability, which may play an important role in susceptibility to IBD, has been observed in patients with both asthma [35] and COPD [36], further substantiating the connection between the gut and chronic airway diseases. Moreover, activities of daily living led to enterocyte damage along with intestinal hyperpermeability in patients with COPD, but not in control subjects, indicating functional alteration in the gastrointestinal tract [36]. These findings provide evidence of an intestinal compromise and an associated mechanism for intestinal dysfunction in patients with COPD, not yet sufficiently identified.
The potential genetic, physiological and immune mechanisms underlying the connections between IBD and chronic airway diseases have been extensively covered by Keely et al. [37]. Four mechanisms have been invoked in this so-called lung–gut cross-talk [38] (figure 2).

Potential mechanisms involved in lung–gut cross-talk. Inflammation may lead to the production of autoimmune antibodies against the elastin or autoimmune responses against antigens produced during cigarette smoke-induced oxidative stress. Systemic interleukin (IL)-6, along with localised transforming growth factor (TGF)-β may drive cross-organ T-helper cell 17 polarised inflammation. Systemic IL-13 may drive aberrant natural killer T-cells and macrophage responses across organs. CytOx: cytochrome oxidase; DC: dendritic cells. Reproduced from [37] with permission from the publisher.
First, an overproduction of proteases has been identified in COPD and in animals [39], namely the matrix metalloproteinase (MMP) family of proteases with a role in the digestion of key components of mucosal structural integrity [39, 40]. Increased levels of the proteases that break down connective tissue components have been identified in patients with COPD and modelled in animals [39]. It has been shown that the MMP family of proteases has a role in the digestion of collagen, elastin, fibronectin and gelatin, vital constituents of the mucosal integrity of the lung [40]. Increased levels of epithelial and leukocyte MMP-2, MMP-9 and MMP-12 have been associated with the pathogenesis of COPD [39, 41, 42] and IBD [43, 44], thus contributing to a remodelling process. Despite the well established role of α1-antitrypsin (α1-AT), an enzyme with serum trypsin inhibitor activity and its deficiency in COPD, the prevalence of α1-AT in IBD is questionable [38] and requires further investigation.
Second, both COPD and IBD are considered to be driven by low-grade chronic systemic inflammation so that circulating lymphocyte activity may be a contributory factor [45–47]. During inflammation, bronchial lymphoid tissue is regulated by lymphocyte traffic from the lung tissue through the general circulation [48]. This mirrors the role of the intestinal lymphoid tissue so that both lung and intestinal lymphocytes migrate to other mucosal sites as part of the common mucosal immune system. This traffic may be responsible for the presence of systemic inflammation associated with both COPD and IBD. Increased cytochrome oxidase activity has been observed in circulating lymphocytes of patients with COPD [49], a response that has also been observed in other chronic inflammatory disorders, such as bronchial asthma and rheumatoid arthritis, but whether these same responses occur in IBD remains unknown. Cigarette smoke also seems to affect the homing properties and maturation of dendritic cells [50, 51], considered to be vital as antigen-presenting cells in mucosal immune responses.
Third, an abnormal lung microbiome has been reported in patients with COPD [52] and it is known that smoking restricts the ability of alveolar macrophages to phagocytose and kill bacteria [53], suggesting that cigarette smoke may lead to an immune defect of the lung microbiome. There is evidence that Gram-negative bacteria of the enteric microflora may also be part of the lung microbiota [54, 55], micro-organisms that are resistant to cigarette smoking [56] and may contribute to severe exacerbations of COPD [55]. Furthermore, an inappropriate immune response against intestinal microflora is now widely accepted to play a critical role in the inflammation process associated with IBD. Thus, there exists the possibility that an immune response against micro-organisms in intestinal microflora observed in IBD may also be directed toward enteric bacteria of the lung microbiome.
Fourth, there is evidence to suggest that COPD has an autoimmune component, which can lead to progression and relapse of disease [57, 58]. This is consistent with the concept that both the activity and severity of the natural history of COPD continue and progress even after the cessation of smoking, thus suggesting that an immune response occurs against components other than cigarette smoke. Smoke-induced emphysema has been shown to generate an autoimmune response against elastins [59, 60]. Teleologically, Crohn's disease is considered to be a disease with an autoimmune background so that its development would be attributed to an initial infection or trigger leading to an inappropriate immune response against the intestinal mucosa and/or commensal bacterial population [61, 62]. This modulates the characteristic recurring cycles of chronic inflammation in Crohn's disease. Likewise, ulcerative colitis has an autoimmune component different to that of Crohn's disease [63, 64] in that isoforms of human tropomyosin, capable of inducing autoantibodies and T-cell responses, have been observed in ulcerative colitis [65]. Similarly, autoimmunity could explain some elements of organ cross-talk in inflammatory disease. Immune responses against bacteria or conserved mucosal protein epitopes of the pulmonary and gastrointestinal tracts may explain cross-organ inflammation in COPD and IBD.
Given the limited research to date, further comprehensive studies on the prevalence of intestinal involvement in COPD and of pulmonary diseases among IBD patients are warranted. The mechanisms that highlight the development of systemic inflammation in IBD and COPD patients are confounded by the complex aetiologies of these conditions that can share environmental triggers and have similar immune and physiological involvement.
Chronic liver diseases
In this section, we focus on the lung–liver cross-talk. Here, all sorts of chronic respiratory and liver diseases are highlighted, although we clearly prioritise the predominance of liver-induced pulmonary vascular diseases, namely POPH and HPS. Likewise, we refer to the most common lung function abnormalities in chronic hepatic diseases, including the potential beneficial role played by increased serum bilirubin levels on lung function in chronic respiratory diseases. This is followed by a more detailed description of both POPH and HPS and the interplay between a variety of chronic liver disorders and a heterogenous group of chronic respiratory diseases.
Lung function abnormalities in liver diseases
In parallel to obstructive and restrictive spirometric abnormalities, the most common lung function disturbance in patients with advanced liver disease is a decreased TLCO [66]. The coexistence of considerable ascites and fluid retention can alter both lung mechanics and airways. Together these pathophysiological disurbances reduce lung volumes and minimise the efficiency of pulmonary gas exchange, resulting in abnormally increased alveolar–arterial oxygen tension difference (PA–aO2) with or without arterial hypoxaemia, associated with normal or low arterial carbon dioxide tension [67, 68].
Several studies [69–71] have indicated that normal bilirubin serum levels may reflect a combination of environmental and genetically determined susceptibility to respiratory diseases so that bilirubin may have a favourable effect on lung function in patients with COPD. Similar to earlier studies [72–74], current smoking and its duration, higher body mass index and higher levels of social deprivation were all associated with moderately lower concentrations of bilirubin. Using data from the Lung Health Study [70], it has recently been observed that serum bilirubin was positively related to lung function outcomes and rate of progression of airflow limitation in smokers with mild-to-moderate COPD, independently of cigarette smoking and other risk factors [75] (figure 3). These findings may indicate that bilirubin levels appear to be depleted due to exposure to reactive oxidative species, such as those derived from cigarette smoking [72, 76]. The degradation of haem to biliverdin by haem-oxygenase (HO), where the production of bilirubin originates, results in biliverdin being subsequently reduced to bilirubin by biliverdin reductase [77]. Moreover, HO-1, the inducible isoform of HO, upregulated by oxidative stress [78], is expressed increasingly in hypoxia and in type II pneumocytes and alveolar macrophages within the lung [79]. Genomic studies have revealed that the relative gene expression of HO and biliverdin reductase is increased in the lung [80]. The increased HO-1 expression in an experimental model attenuated the severity of emphysema [81], while the HO-1 gene contained a variable number of gene giant (GT) nucleic acid repeats. Individuals with fewer GT repeats have higher serum levels of bilirubin [82] and a lower risk of COPD [83]. It is known that oxidative stress is generated endogenous and exogenously in COPD. In human lungs, lipid peroxidation causes damage to multiple cell membrane components and impairs cell structure and permeability [84]. Altogether, these studies point to a beneficial protective role of increased serum bilirubin levels on lung function in COPD by inhibiting lipid peroxidation, a mechanism potentially relevant for the prevention and therapy of chronic respiratory diseases [85, 86]. The potential influence of these favourable effects of serum bilirubin in patients with liver-induced pulmonary vascular diseases remains unknown. The abnormally decreased TLCO frequently observed in advanced liver diseases, including HPS, remains an intriguing finding and is still incompletely resolved. A diffusion–perfusion defect [87] was originally implicated in the oxygen diffusion gradient in dilated pulmonary vessels. The pronounced pulmonary vasodilatation at the capillary or precapillary level in cirrhosis would cause substantial ventilation–perfusion (V′/Q′) imbalance, mainly characterised by regions of low or extremely low V′/Q′ ratios along with mild-to-moderate intrapulmonary shunt. These two abnormal traits represent the key intrapulmonary determinants governing pulmonary gas exchange disturbances in HPS [88]. In addition, it has been suggested that there may be inadequate diffusion of oxygen to the centre of the enlarged capillaries in the lung, resulting in partially deoxygenated blood. The achievement of complete alveolar–capillary equilibrium for oxygen also depends on the transit time of the red blood cell. The inordinately high cardiac output conspicuously present in patients with advanced liver diseases, most commonly observed in liver cirrhosis, resulting in a shorter transit time would further aggravate the limitation of diffusion by this mechanism.

Plots of post-bronchodilator forced expiratory volume in 1 s (FEV1) decline and serum bilirubin levels between years 2 (at baseline) and 5 (time of blood sampling). a) Univariate relationship; b) the relationship statistically adjusted for age, sex, body mass index, race, post-bronchodilator FEV1 at baseline, logarithm of methacholine reactivity and pack-years smoked. Reproduced from [75] with permission from the publisher.
Pulmonary vascular-induced liver disorders
There are two salient liver-induced pulmonary vascular disorders that deserve to be considered under this subheading: POPH and HPS [89–92]. Both are increasingly recognised due to the therapeutic success of liver transplants [93]. Moreover, the coexistence of these two hepatopulmonary vascular syndromes affects both survival and indications for liver transplant. Currently, the prevalence of POPH is of the order of 5% in chronic liver patients submitted to liver transplant; however, this surgical approach is contraindicated in POPH given its negative peri- and post-operative impact. In contrast, liver transplant is the only effective treatment for improving outcome in patients with HPS, a life-threatening condition whose prevalence can be close to 20% in patients awaiting liver transplant. Both POPH and HPS are associated with portal hypertension and/or liver disease. It can be succinctly stated that POPH is related to vasoconstriction and/or vascular obstruction in pulmonary vessels while HPS is linked to excessive pulmonary vasodilatation. Furthermore, a specific common pulmonary vascular phenotype has been suggested given their rare simultaneous or sequential coincidence in the same patient. As a very recent example of this potential interplay, pulmonary vascular dilatation, an essential feature of HPS but not typically characteristic of POPH, has been frequently reported in POPH along with decreased survival, an intriguing observation that warrants further research [94]. Nonetheless, these two liver-induced pulmonary vascular conditions encompass a distinct pathobiology, as well as different management and treatment, which need further research.
Current status of POPH
POPH is defined as a pulmonary arterial hypertension (PAH) associated with portal hypertension [90, 95–97]. Diagnosis of POPH is based on pulmonary haemodynamic criteria obtained via right heart catheterisation [97–99]. PAH includes other forms of precapillary pulmonary arteriopathy, such as idiopathic (formerly “primary”) (i)PAH and heritable PAH, as well as forms associated with other triggers (i.e. medication or toxin use, congenital heart disease, connective tissue disease or HIV infection).
POPH has been reported in 0.13% of nonselected patients versus 0.73% of patients with cirrhosis and portal hypertension [100]. Clinical series with proven cirrhosis showed a prevalence of PAH in the range 0.61–2.0% [100, 101]. A prospective study of >1000 patients evaluated for liver transplant showed that 5% met current haemodynamic criteria for POPH [102], a figure close to those reported by prior retrospective studies [103, 104]. It is of note that the correlation between aetiology of liver disease, degree of portal hypertension, systemic haemodynamics and severity of PAH remains poor [99, 101, 105, 106]. POPH accounts for ∼5–10% of patients with PAH in France and the United States [107–109]. A 2012 USA-based PAH registry has suggested that, despite similar functional class, patients with POPH display less severely impaired haemodynamics at presentation compared with patients with iPAH [110].
Co-infection associated with HIV is a risk factor for PAH, and chronic hepatitis C virus (HCV) infection may have unique synergistic effects on the pulmonary vasculature. The estimated prevalence of PAH in patients with HIV is ∼0.5% [111]. In a recent study, a preliminary prevalence of echocardiographic PAH in HIV–HCV was considerably higher than that reported for HIV alone. Moreover, time since HCV diagnosis and treatment of HCV with interferon were both significantly associated with the development of PAH and right ventricular dysfunction as assessed by echocardiography [112]. Accordingly, the prevalence of echocardiographic PAH may be higher in HIV–HCV co-infected patients than in those with HIV mono-infection alone, a contention that deserves further investigation as potential aetiology of pulmonary vascular disease.
The mechanisms of other types of PAH may not necessarily be the same as those underlying POPH [90]. Expression of prostacyclin synthase appears to be reduced within pulmonary arteries in both subsets of PAH [113], while endothelin abnormalities have been identified in iPAH and liver cirrhosis and portal hypertension [114]. Compared with patients without POPH, those with POPH and refractory ascites have greater endothelin-1 levels in pulmonary arteries [115]. Genetic variants of both aromatase and oestrogen receptor-1 and single nucleotide polymorphisms in some genes have been associated to the risk of POPH [116].
The immediate goal of pulmonary vasomotor pharmacological therapy for POPH is to improve pulmonary haemodynamics by reducing the obstruction to pulmonary arterial flow. This can be approached with medications that selectively or in combination result in vasodilatation, antiplatelet aggregation and antiproliferative effects [117]. The development of compounds that specifically target pathways involved in the pathogenesis of PAH has been associated with better quality of life and improved clinical outcomes in patients affected with PAH. These goals can be achieved through several pathways: by augmenting pulmonary endothelial prostacyclin synthase deficiency, blocking circulating endothelin-1 effects with endothelin receptor antagonists and enhancing local nitric oxide vasodilatation effects using PDE inhibitors. The ultimate target is to stabilise, improve or normalise right ventricular function along with improving beneficial pulmonary haemodynamics. Nonetheless, the outcome of POPH following liver transplant remains unsettled despite selective current substantial advances in single and combination pulmonary vasomodulatory therapies.
Co-occurrence of HPS and chronic respiratory diseases
HPS is characterised by a clinical triad that encompasses a defect in arterial oxygenation induced by pulmonary vascular dilatation in the setting of liver disease [88, 89]. A classification of the severity of the HPS based on abnormalities in oxygenation is vital because severity critically influences survival and is useful in determining the timing and risks of liver transplant [88]. In 2004 the European Respiratory Society task force defined HPS by the presence of 1) liver disease (usually chronic); 2) abnormal arterial oxygenation (defined as PA–aO2 >15 mmHg (or >20 mmHg in those aged >65 years) or arterial oxygen tension (PaO2) <80 mmHg while breathing ambient air) in the absence of an alternative cause; and 3) evidence of pulmonary vascular dilatation, most commonly diagnosed using contrast transthoracic echocardiography [88].
The simultaneous measurement of both TLCO and transfer factor of the lung for nitric oxide in patients with hepatic diseases, including HPS, has suggested that a decreased lung capillary blood volume and reduced thickness of the capillary blood layer along with a reduced transit time of the red blood cells in the lungs are the most plausible mechanisms for decreased TLCO [118]. These changes result from decreased precapillary arteriolar tone that coexists with a pulmonary vasculature that behaves paradoxically by combining a lower hypoxic vascular response along with reversion of hypoxic pulmonary vasoconstriction. Although no explanation for these findings has yet emerged, these findings may be consistent with widespread inadequate pulmonary vascular reactivity, ultimately involving additional mechanisms including angiogenesis, vascular remodelling and vasculogenesis [119, 120], a compelling concept that has emerged and gathered strength over the past years.
Along these lines it has been observed that pulmonary exchange disturbances in HPS remain unchanged after acute nebulisation of NG-nitro-l-arginine methyl ester, an inhibitor of nitric oxide synthase [119]. This suggests that HPS-induced gas exchange disturbances may be related to pulmonary vascular remodelling and/or vasculogenesis rather than to an ongoing vasodilator effect of enhanced nitric oxide production [121]. Single nucleotide polymorphisms in genes related to angiogenesis, such as endoglin, endostatin, angiopoietin-1 and TIE (tyrosine kinase with immunoglobulin-like and endothelial growth factor-like domains)-1 were associated with the presence of HPS in patients with portal hypertension [116]. Enhanced pulmonary production of nitric oxide has been implicated in the development of pulmonary vascular dilatation in cirrhotic patients with HPS [121–126]. Exhaled nitric oxide levels are increased in HPS patients and normalise after liver transplant, as the impact of HPS resolves or minimises. However, the mechanisms of increased endogenous nitric oxide production and their relationship to the presence of portal hypertension, the hyperkinetic circulation and the degree of liver injury, remains unknown. In addition, whether other mediators might contribute to pulmonary vascular dilatation is a contention that has not yet been sufficiently investigated.
Data from liver transplant centres indicate that the prevalence of HPS, including that involving mild categories, ranges from 5% to 32% [127]. No prospective multicentre prevalence studies have been reported to date. The range in prevalence is primarily a function of varying abnormal PA–aO2 and PaO2 cut-offs that are used to define gas exchange disturbances and grade clinical severity of HPS [88, 89, 127].
Multimorbidities such as cardiovascular disease, COPD and pulmonary fibrosis may be coexisting conditions in many patients with liver disease, a situation that can confound the clinical characteristics of HPS and interfere with pulmonary gas exchange disturbances [88, 128, 129]. Accordingly, the finding of an abnormally increased PA–aO2 with or without arterial hypoxaemia should prompt a methodological systematic approach to identify alternative contributing respiratory conditions, using thoracic computerised tomography to rule out obstructive and/or restrictive respiratory diseases [89, 90]. It is difficult to definitively diagnose HPS in patients with significant coexistent lung disease, as there is no specific test to differentiate gas exchange abnormalities due to HPS from those due to other pulmonary disorders in the presence of pulmonary vascular dilatation. Compared with cirrhotic patients without HPS, those with HPS have a lower TLCO, but this functional disturbance is not sufficiently discriminative [129].
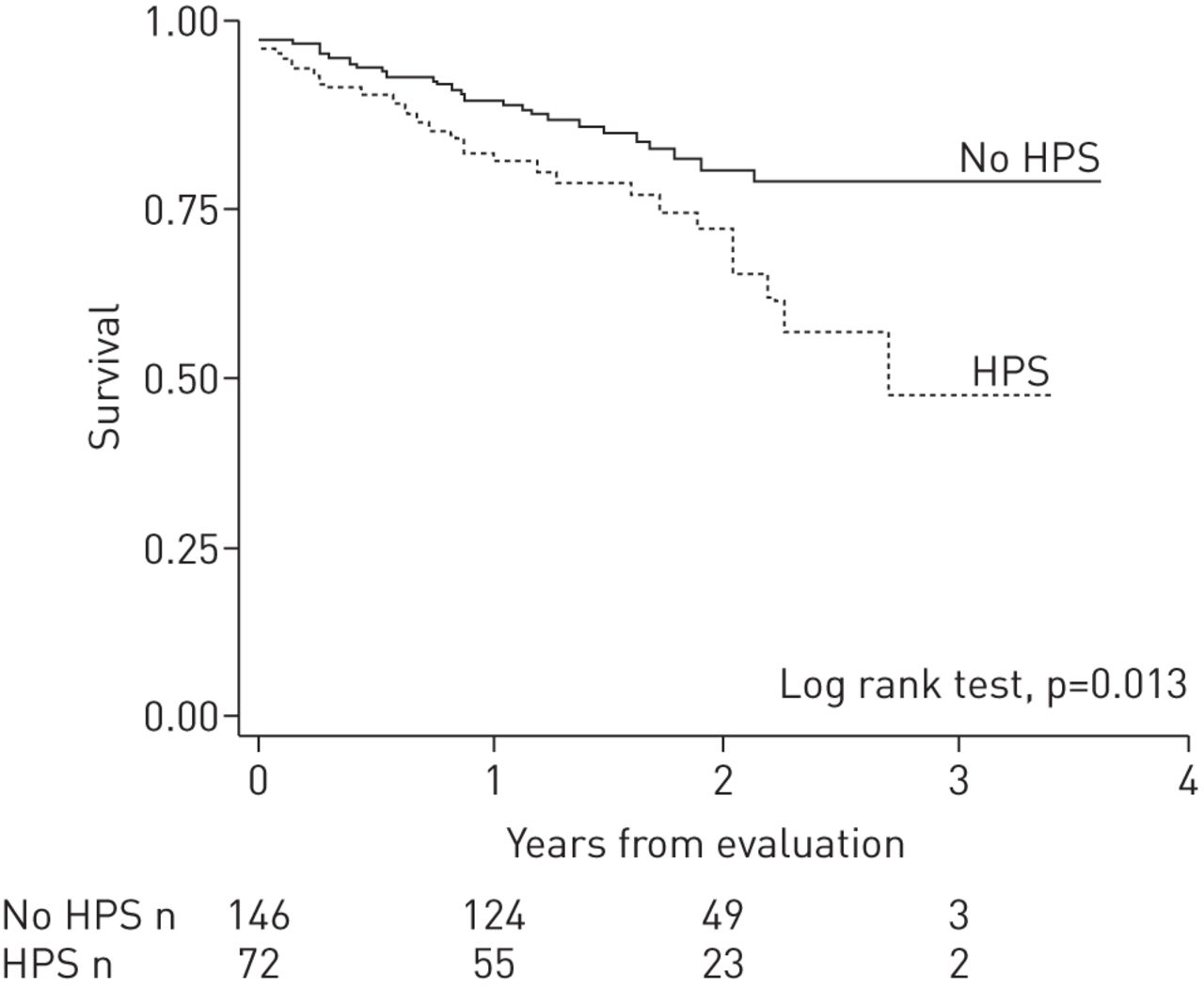
Patients with HPS have significantly worse functional class and more impaired quality of life than those without HPS even after adjustment for age, sex and severity of liver disease [129]. In this prospective multicentre study of candidates for liver transplant, patients with and without HPS had a low prevalence of COPD (7% and 3%, respectively) [129]; however, HPS was more common in lifelong nonsmokers. The presence of HPS in liver transplant candidates represented a doubled risk of death compared with those without HPS, even after adjustment for potential confounders [129] (figure 4). Similarly, patients with HPS have a significantly lower 5-year survival rate (0–23%) compared with that of patients with advanced liver disease without HPS (30–60%) in the absence of liver transplant [130, 131].
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Kaplan–Meier survival estimates of patients with hepatopulmonary syndrome (HPS) and without HPS (n=218) from a prospective multicentre cohort study of patients evaluated for liver transplant. Patients with HPS, compared with those without HPS, exhibited a two-fold increase of the risk of death. The median time from diagnosis of liver disease to evaluation for transplant (entry to the study) was 3 years (range 1–11 years) for patients with HPS and 4 years (range 1–9 years) for those without HPS. Reproduced from [129] with permission from the publisher.
Patients with HPS awaiting liver transplant exhibit an average decline in PaO2 of ∼5 mmHg year-1 [90]. Some studies have suggested that very severe arterial hypoxaemia in HPS confers a worse prognosis with or without liver transplant [129, 131, 132], an observation that has not been replicated in all series [133]. Patients with HPS frequently die of complications of liver disease, namely hepatic failure, gastrointestinal haemorrhage, sepsis or infection, and hepatorenal syndrome, all complications of liver disease [90], rather reflecting that HPS represents a systemic disorder with implications that extend beyond pulmonary or hypoxaemia related events.
HPS may coexist with other cardiorespiratory disorders and contributes significantly to abnormal pulmonary gas exchange abnormalities under these circumstances [129]. Although it has been anticipated that HPS and coexistent chronic pulmonary diseases, such as COPD or pulmonary fibrosis, can occur in up to 30% of candidates to liver transplant, the evidence is still weak and needs to be further substantiated [128]. Notably, the distinctively unique features of pulmonary haemodynamic and gas exchange hallmarks in patients with HPS do not seem to be aggravated by the coexistence of chronic cardiorespiratory conditions, a finding with clinical relevance for elective indication of liver transplant in patients with HPS that warrants further investigation. Furthermore, a very recent investigation identified HPS as a common cause of dyspnoea in patients with the short telomere syndrome [92, 134].
While no effective pharmacological and nonpharmacological therapy for the HPS exists, liver transplant has become the only successful therapy [135]. However, both post-operative mortality and the interval between transplant and the resolution of arterial hypoxaemia are increased in patients with severe arterial deoxygenation [136].
Other chronic hepatic diseases
Chronic active hepatitis, a frequent chronic liver disease characterised by diffuse parenchymal inflammation and hepatic cell necrosis, may be caused by viral hepatitis (most commonly HCV), autoimmune disorders and drug-related liver injury. Pulmonary fibrosis and lymphoid interstitial pneumonitis have been reported but are rare [137]. Chronic active hepatitis can lead to liver cirrhosis and hepatic failure, which has been associated with HPS [138]. Patients with chronic HCV infection and coexistent COPD may demonstrate an accelerated annual decline in forced expiratory volume in 1 s and TLCO [139].
Primary biliary cirrhosis (PBC) is an autoimmune disease, characterised by a chronic, cholestatic, granulomatous and destructive process that involves the intrahepatic bile ducts. More severe involvement result in cholestasis, cirrhosis and liver insufficiency [140]. The association of immunological abnormalities, such as depressed T-suppressor cell function, hypergammaglobulinaemia and anti-mitochondrial antibodies reflects its autoimmunological basis. Sicca complex, Sjögren's syndrome and scleroderma are frequently associated with PBC [141], which also may be associated with POPH [106]. Several respiratory abnormalities have been associated with PBC, including interstitial lung disorders, such as lymphocytic interstitial pneumonitis and pulmonary fibrosis, subclinical intrapulmonary granulomas that mimic sarcoidosis [142] and widespread bronchiectasis [138].
Conclusion
There is solid evidence of a connection between the lung and the gastrointestinal tract, on the one hand, and the liver, on the other, in adult patients. The lung–gut cross-talk refers to the subset of IBD, namely Crohn's disease and ulcerative colitis, and its well-known association with a heterogenous spectrum of respiratory multimorbitidies, as well as the increased prevalence and incidence of IBD in patients with chronic airways diseases, such as COPD and bronchial asthma. Furthermore, there is a lot of knowledge about the potential shared mechanisms of the two systems involved. A huge research effort is still warranted to improve our understanding and further identify the underlying mechanisms. Alternatively, the lung–liver interplay is predominantly centred on the progressively increased incidence of hepatopulmonary vascular disorders, namely POPH and HPS, along with a variety of other chronic respiratory disorders coexisting with chronic hepatic diseases. The knowledge of the mechanisms of liver-induced pulmonary vascular entities has progressively improved and grown, where the underlying mechanism of angiogenesis is emerging. In the light of the big impact that disorders of gut and liver have on the lung, internal medicine and respiratory physicians should increase awareness of these multimorbidities to identify them and improve the management and therapy of these patients.
Footnotes
Previous articles in this series: No. 1: Faner R, Cruz T, López-Giraldo A, et al. Network medicine, multimorbidity and the lung in the elderly. Eur Respir J 2014; 44: 775–788. No. 2: Divo MJ, Martinez CH, Mannino DM. Ageing and the epidemiology of multimorbidity. Eur Respir J 2014; 44: 1055–1068. No. 3: MacNee W, Rabinovich RA, Choudhury G. Ageing and the border between health and disease. Eur Respir J 2014; 44: 1332–1352. No. 4: Carraro S, Scheltema N, Bont L, et al. Early-life origins of chronic respiratory diseases: understanding and promoting healthy ageing. Eur Respir J 2014; 44: 1682–1696. No. 5: Chacko A, Carpenter DO, Callaway L, et al. Early-life risk factors for chronic nonrespiratory diseases. Eur Respir J 2015; 45: 244–259. No. 6: Barnes PJ. Mechanisms of development of multimorbidity in the elderly. Eur Respir J 2015; 45: 790–806.
Conflict of interest: Disclosures can be found alongside the online version of this article at erj.ersjournals.com
- Received April 24, 2015.
- Accepted November 12, 2015.
- Copyright ©ERS 2016
References










