





Abstract
Pulmonary hypertension can be caused by obliterative phlebitis in patients with IgG4-related lung disease http://ow.ly/EEjvE
To the Editor:
IgG4-related disease (IgG4-RD) is a systemic fibroinflammatory disease associated with elevated serum IgG4 level and IgG4-positive plasma cell infiltration into multiple organs [1]. In 2001, Hamano et al. [2] reported that patients with autoimmune pancreatitis have high-serum IgG4 concentrations. Subsequently, IgG4-related lesions similar to those of autoimmune pancreatitis have been identified in other organs [1] including the lungs [3].
Thoracic involvement in IgG4-RD has been increasingly recognised, usually manifesting as inflammatory pseudotumours, interstitial pneumonia or occasionally, pleuritis [3]. However, little information is available on airway disease or pulmonary hypertension (PH) associated with IgG4-RD. In this report, we describe a patient presenting with PH associated with IgG4-related bronchiolitis.
A 45-year-old male first presented with community-acquired pneumonia caused by Haemophilus influenzae. When he was discharged, slight dyspnoea on exertion and abnormal findings in computed tomography (CT) of the chest remained. After 5 months of follow-up, he was admitted again for a close inspection of deteriorating dyspnoea, which was categorised as World Health Organization class II. He had a 10-year history of refractory sinusitis. For the last 5 years, he had complained of swelling of the bilateral eyelids, which was diagnosed as “nonspecific dacryoadenitis” based on left lacrimal grand biopsy at a previous hospital clinic visit. He was an ex-smoker (one pack per day for 20 years) and had no relevant occupational or environmental exposures.
A chest CT scan manifested diffuse bilateral centrilobular nodules, prominent peripheral pulmonary vascular markings (fig. 1a) and mediastinal lymphadenopathy with no evidence of pulmonary large vessel compression (fig. 1b). Laboratory examinations showed high serum IgG (6530 mg·dL−1) and IgG4 (>1500 mg·dL−1) levels. Serum C-reactive protein, lactate dehydrogenase, Krebs von den Lungen-6 antigen and surfactant protein-D levels were normal. Results of evaluations for connective tissue diseases, angiotensin converting enzyme, human leukocyte antigen-B54 and human T-lymphotropic virus-1 antigen were all negative. Arterial blood gas showed normal partial pressure of oxygen and carbon dioxide (87.5 and 35.3 Torr, respectively). A pulmonary function test indicated obstructive disorder with decreased forced expiratory volume in 1 s (FEV1)/forced vital capacity ratio (FVC) and decreased predicted value for diffusing capacity of the lung for carbon monoxide (%DLCO) (FEV1/FVC 67.3%; %DLCO 73.6%). The predicted values for FVC (%FVC) and FEV1 (%FEV1) were normal (%FVC 108.4%; %FEV1 81.6%). Bronchoalveolar lavage fluid cell count was 0.32×105 cells·mL−1, with a normal cellular profile and CD4/CD8 ratio. Microbial culture results on the sputum and bronchoalveolar lavage fluid were negative. Although echocardiography disclosed no abnormalities, pulmonary arterial catheterisation revealed elevated pulmonary arterial pressure (PAP) (systolic 47 mmHg, diastolic 23 mmHg, mean 33 mmHg) and elevated pulmonary vascular resistance (PVR) (4.47 Wood units), with normal right atrial pressure (RAP) (7 mmHg), cardiac index (3.07 L·min−1·m−2) and pulmonary arterial wedge pressure (PAWP) (9 mmHg). 6-min walk distance (6MWD) was 648 m, with 91% of minimum arterial oxygen saturation measured by pulse oximetry (SpO2).

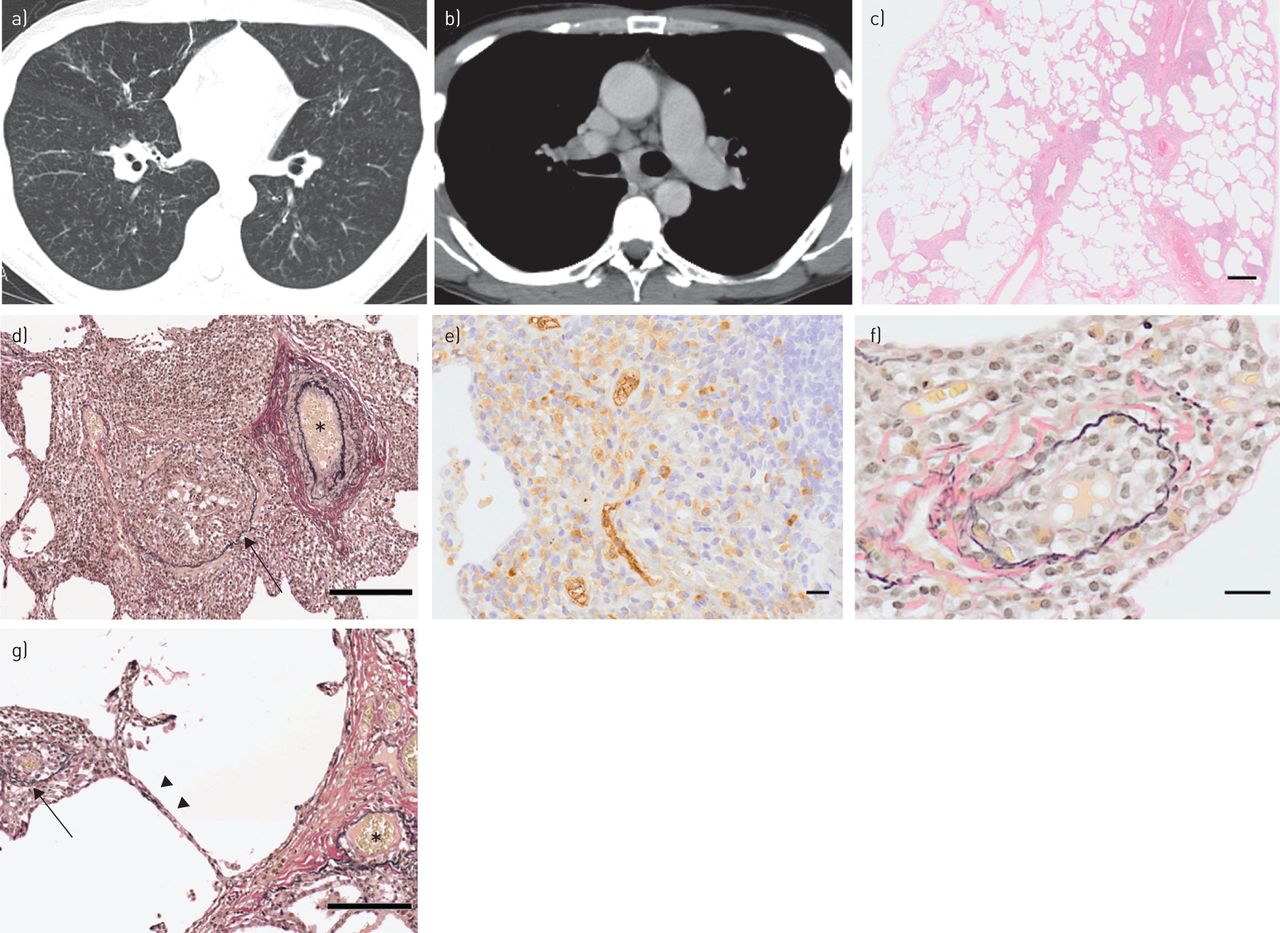{kind=link}
Radiological findings on chest computed tomography (CT) scan and histopathological findings in lung biopsy specimen. a) Chest CT scan showing diffuse bilateral centrilobular micronodules and prominent peripheral pulmonary vascular markings. b) Contrast-enhanced chest CT scan showing mediastinal lymphadenopathy with no evidence of pulmonary large vessel compression. c) Photomicrograph showing intense inflammation expanding from bronchioles into surrounding lung parenchyma (haematoxylin–eosin stain). d) Elastica van Gieson (EVG) stain showing constrictive bronchiolitis (arrow) and uninvolved artery (asterisk). e) IgG4 immunohistochemical stain showing a remarkably high number of IgG4-positive plasma cells per high-power field and an IgG4/IgG ratio of ∼50%. f) EVG stain showing obliterative phlebitis with plasma cells. g) EVG stain showing obliterative phlebitis (arrow), uninvolved artery (asterisk) and adjacent alveolar septa with uninvolved capillaries (arrowheads). Scale bars: 500 μm (c), 200 μm (d and g) and 20 μm (e and f).
Suspecting IgG4-related lung disease, we performed video-assisted thoracoscopic surgery. The specimen exhibited marked peribronchiolar inflammation due to infiltrating plasma cells, lymphocytes and eosinophils. Some foci showed expanded infiltration into the surrounding lung parenchyma (fig. 1c and d), which corresponded with the diffuse bronchiolocentric nodules seen on chest CT. Immunostaining showed 30 IgG4-positive plasma cells per high-power field and an IgG4/IgG ratio of 50% (fig. 1e). Nearly half of the medium-sized veins were involved through lymphoplasmacytosis, and a small number of veins among them showed destruction of the lamina elastica in their walls. These veins were considered to show obliterative phlebitis (fig. 1d, f and g). No inflammation of the pleura and thrombus, and no veno-occlusive changes, capillary hemangiomatosis, capillaritis or obliterative arteritis were observed (fig. 1d and g). After confirming these findings, we obtained a lacrimal gland specimen from the previous hospital, in which notable infiltration of IgG4-positive plasma cells was observed with an IgG4/IgG ratio of >50%. Diagnoses of IgG4-related bronchiolitis and dacryoadenitis were made, and the cause of PH was guessed to be obliterative phlebitis.
6 months after surgical biopsy, dyspnoea, eyelid swelling and postnasal discharge had worsened. %FEV1 declined to 47%. Right heart catheterisation showed elevated PAP (systolic 58 mmHg, diastolic 28 mmHg, mean 42 mmHg) and elevated PVR (5.18 Wood units) with normal RAP (8 mmHg), cardiac index (3.41 L·min−1·m−2) and PAWP (11 mmHg). 6MWD decreased to 555 m with a minimum SpO2 of 85%. Based on these findings, two cycles of methylprednisolone pulse (1000 mg·day−1 for 3 days) were initiated, followed by 10 mg of prednisolone and cyclosporin daily on the basis of the authors' experience with collagen tissue disease-related interstitial pneumonias [4]. This was associated with significant resolution of symptoms within 1 month without recurrence or any adverse events.
After 3 months of treatment, the abnormal CT findings had markedly improved. Serum IgG4 level decreased to 358 mg·dL−1, while %FEV1 and %DLCO increased to 89.0% and 96.9%, respectively. A repeat pulmonary arterial catheterisation showed a decline in PAP (systolic 34 mmHg, diastolic 13 mmHg, 23 mmHg) and a decline in PVR (2.30 Wood units) with normal RAP (3 mmHg), cardiac index (4.58 L·min−1·m−2) and PAWP (5 mmHg). 6MWD and minimum SpO2 were improved to 705 m and 90%, respectively.
IgG4-RD is a novel clinical disease entity characterised by elevated serum IgG4 concentration and marked infiltration of lymphocytes and IgG4-positive plasma cells in various organs [1]. In this patient, the diagnosis was confirmed by a serum IgG4 level well above the cut-off value for diagnosis (135 mg·dL−1) and by the typical lymphoplasmacytic infiltrates containing IgG4-positive plasma cells (>10 IgG4 cells per high-power field and an IgG4/IgG cell ratio >40%) [5] in biopsy specimen.
Inoue et al. [6] reported two cases of IgG4-related lung disease with thickening of bronchovascular bundles and multiple centrilobular nodules on chest CT, which histologically corresponded to inflammation in the peribronchiolar area. Although our case did not show thickening of bronchovascular bundles on chest CT, other findings corresponded with these two cases. Other typical morphologic features of IgG4-RD, such as obliterative phlebitis and mild-to-moderate eosinophil infiltration, were also observed in this patient. Obliterative phlebitis is less commonly observed in IgG4-related lung diseases other than those with pseudotumour and pleuritis phenotypes [7, 8]; however, this case showed remarkable obliterative phlebitis.
IgG4-RD usually presents in the lungs with pseudotumours, interstitial pneumonia or pleuritis [3], while airway involvement is an unusual phenotype. There are a few articles available about central airway disease, such as bronchial stenosis [9] or bronchiectasis caused by direct infiltration of IgG4-positive cells around bronchovascular bundles with or without bronchiolitis [6, 7, 10]; however, we could only find two articles that focussed mainly on IgG4-related bronchiolitis [11, 12]. Matsui et al. [11] first described a case with IgG4-related bronchiolitis associated with Mikulicz's disease and autoimmune pancreatitis. Subsequently, Hasegawa et al. [12] reported a patient with IgG4-related bronchiolitis and bronchial asthma. Both of these cases were diagnosed by surgical lung biopsy and improved with steroid therapy.
Ishida et al. [13] reported the first case of PH associated with IgG4-RD. Although the patient underwent transbronchial lung biopsy, the IgG4/IgG cell ratio was not elevated in the specimen and they were unable to prove the aetiology of PH histologically. Consequently, this is the first case report to provide histological evidence of PH associated with IgG4-related lung disease. In this case, PH was considered to be caused by obliterative phlebitis from IgG4-positive plasma cells, which was hemodynamically similar to pulmonary veno-occlusive disease. Pulmonary veno-occlusive disease is classified as a subgroup of pulmonary arterial hypertension with the observed post-capillary lesions of septal veins and pre-septal venules consisting of loose, fibrous remodelling of the intima that may totally occlude the lumen [14]. This case can be distinguished from pulmonary veno-occlusive disease by its completely different pathological findings.
Mediastinal lymphadenopathy or fibrosing mediastinitis, which are sometimes caused by IgG4-RD [1], can cause pulmonary arterial or venous compression [15] and should also be differentiated from obliterative phlebitis as an aetiology of PH. In this case, contrast-enhanced chest CT showed no evidence of pulmonary large vessel compression.
In conclusion, this is the first report describing histological evidence of PH associated with IgG4-RD, which was improved by steroid and cyclosporine therapy. Pulmonary artery catheter and lung biopsy should be considered in cases of unexplained dyspnoea and bronchiolitis. Further studies are warranted to investigate the spectrum of IgG4-RD.
Footnotes
Conflict of interest: None declared.
- Received July 21, 2014.
- Accepted November 15, 2014.
- Copyright ©ERS 2015
References










